АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомДругоеЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция
ХII.2. Гомогенний каталіз
|
Читайте также: |
У гомогенному каталізі каталізатор та речовини, що реагують, знаходяться в одній фазі. Найбільш часто такий вид каталізу зустрічається в розчинах. Основний механізм дії гомогенного каталізатора - це утворення реакційноздатних проміжних сполук.
 Зниження енергії активації для найпростішої реакції за участю каталізатора в порівнянні з некаталітичною реакцією показано на рис. 69. Механізм каталітичної реакції можна подати таким чином:
Зниження енергії активації для найпростішої реакції за участю каталізатора в порівнянні з некаталітичною реакцією показано на рис. 69. Механізм каталітичної реакції можна подати таким чином:
 |
k1 
 А + К АК (1)
А + К АК (1)
k2
k3
 АК В + К (2)
АК В + К (2)
Перша реакція – це реакція утворення проміжної хімічної сполуки АК при взаємодії вихідної речовини А з каталізатором К (енергія активації Е1). Реакція може бути оборотною. Друга реакція – це реакція розпаду проміжної речовини з утворенням продукту реакції В і відновленням каталізатора (енергія активації Е2).
Сумарна швидкість процесу і концентрація проміжної речовини визначається співвідношенням швидкостей окремих стадій реакцій.
Якщо швидкість другої реакції більша, то загальна швидкість реакції визначається швидкістю першої реакції; концентрація проміжної речовини мала. Швидкість реакції пропорційна концентрації вихідної речовини і каталізатора.
Якщо швидкість першої реакції більша швидкості другої, то загальна швидкість реакції визначається швидкістю другої реакції; концентрація проміжної речовини велика (зв’язаний практично весь каталізатор). Друга реакція не змінює рівноваги, що встановлюється для першої реакції. Цей випадок найбільш цікавий з кінетичної точки зору, а тому розглянемо його більш докладно. Запишемо константу рівноваги для першого процесу

 (XII.2.1)
(XII.2.1)
Тут ми знехтували зміною концентрації вихідної речовини за рахунок утворення проміжного продукту. Знайдемо з цього співвідношення концентрацію сАК:
 (XII.2.2)
(XII.2.2)
Загальна швидкість хімічної реакції запишеться таким чином:
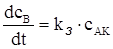 (XII.2.3)
(XII.2.3)
або

 (XII.2.4)
(XII.2.4)
Із одержаної формули, по – перше, видно, що швидкість утворення продукту реакції пропорційна концентрації каталізатора. По – друге, повинен спостерігатись у загальному випадку дробовий порядок реакції. Дійсно, розглянемо граничні випадки:
1) Кс×сА>>1,
тоді  . (XII.2.5)
. (XII.2.5)
Спостерігається нульовий порядок по речовині, що реагує. Це означає, що весь каталізатор зв’язався в проміжний комплекс і швидкість реакції визначається його кількістю.
2) Кс×сА<<1,
тоді  , (XII.2.6)
, (XII.2.6)
спостерігається перший порядок по речовині, що реагує.
Можуть зустрічатися і більш складні випадки. Наприклад, якщо проміжна речовина утворюється з декількох молекул речовини, що реагує, і однієї молекули каталізатора
k1
 nА + K KnA;
nА + K KnA;
k2
k3
 КnA В + К,
КnA В + К,
рівняння за швидкістю реакції запишеться так:
 (XII.2.7)
(XII.2.7)
 Тут порядок реакції може мінятись від 0 до n.
Тут порядок реакції може мінятись від 0 до n.
Іще більш складний випадок спостерігається при утворенні декількох проміжних речовин (наприклад, з різними кількостями молекул речовини, що реагує АК, А2К, А3К та ін.), або коли в утворенні проміжних речовин беруть участь молекули розчинника. Інколи активний комплекс утворюється за участю йонів водню Н+, що присутні в розчині. Тоді швидкість реакції залежить від рН розчину.
Все це приводить до дуже складної залежності швидкості реакції від концентрації речовини, що реагує. Наприклад, для гомогенно-каталітичного розкладу пероксиду водню в присутності солей ванадію або кобальту експериментальна залежність швидкості розкладу Н2О2 від концентрації має вигляд, що показаний на рис. 70.
Причиною такого вигляду кривої вважається утворення двох активних проміжних сполук, які експериментально виділені і визначені. Складна в подібних випадках і залежність кінетики від температури.
Якщо каталітичними властивостями володіють продукти реакції, то такі реакції називають автокаталітичними. В цьому випадку в міру взаємодії концентрація каталізатора зростає, а тому і швидкість постійно зростає.
Всі гомогенно-каталітичні реакції умовно можна розділити на два типи: кислотно-основні і окисно-відновні.
Роль кислот і основ як каталізаторів в реакціях кислотно-основного типу полягає в тому, що вони утворюють з реагентом проміжні сполуки за рахунок донорно-акцепторної взаємодії. При цьому в молекулі проміжної сполуки виникають реакційні центри з підвищеною або зниженою електронною густиною, що веде до полегшення її взаємодії з цим центром (відповідно з електрофільними і нуклеофільними молекулами другого реагента).
В залежності від природи частинок, що виступають у ролі каталізатора, всі каталітичні реакції кислотно-основного типу можна розділити на чотири групи:
1) кислотний каталіз, що включає реакції, які каталізуються лише іонами водню (іонами гідроксонію або ліонію);
2) специфічний каталіз, що включає реакції, які каталізуються іонами гідроксилу;
3) загальний кислотно-основний каталіз, що включає реакції, які каталізуються будь-якими кислотами або основами Бренстеда;
4) електрофільно-нуклеофільний каталіз, що включає реакції, які каталізуються акцепторами або донорами електронних пар, в тому числі і кислотами Льюіса.
Наведемо приклади деяких реакцій кислотно-основного каталізу.
1)Гідроліз складних ефірів у присутності мінеральних кислот включає двосторонню стадію приєднання до карбоксильного кисню за рахунок донорно-акцепторної взаємодії з утворенням карбоній катіона:

Останній, володіючи високою електрофільністю, здатний реагувати з таким відносно слабким нуклеофілом, як вода:

Внаслідок декількох послідовних стадій одержують спирт і кислоту, а також протон, який знову вступає в реакцію.
2) В реакціях специфічного основного каталізу проміжними сполуками є, зазвичай, аніони, які утворюються або приєднанням основи, наприклад ОНˉ , або шляхом відривання протону від реагенту каталізатором. Так протікає альдольна конденсація в присутності ОНˉ-іонів:

Роль протона в активації молекули можуть виконувати деякі електрофільні частинки, в тому числі і кислоти Льюіса. При нуклеофільному каталізі виникають сильно поляризовані молекули проміжної сполуки, що володіють великою реакційною здатністю.
Лімітуючою стадією кислотно-каталітичного процесу, як правило, є перетворення активної проміжної форми в продукти реакції. Константу швидкості k цієї стадії прийнято називати істинною константою швидкості кислотно-каталітичного перетворення. Якщо позначити активну проміжну форму вихідної речовини А через А¹, то швидкість кислотно-каталітичного процесу рівна
 , (XII.2.8)
, (XII.2.8)
якщо А¹ перетворюється мономолекулярно, або
 , (XII.2.9)
, (XII.2.9)
якщо А¹ перетворюється в продукти реакції при взаємодії з молекулою іншої вихідної речовини С. Тут [А¹] – термодинамічно рівноважна концентрація А¹, що складає деяку частку α від сумарної концентрації сА речовини А. В подальшому скрізь для спрощення запису буде передбачатись, що А¹ перетворюється мономолекулярно.
Концентрація А¹ пропорційна сА, тобто α не залежить від сА і визначається лише властивостями середовища. Якщо в ході процесу середовище не змінюється, то можна записати:
 , (XII.2.10)
, (XII.2.10)
деkеф – величина стала на протязі всього процесу. Вона називається ефективною константою швидкості і може бути легко визначена з даних по кінетиці витрачання А відповідними методами. Істинна константа швидкості може бути знайдена, якщо відома частка α активної проміжної форми А¹ речовини А. Концентрація А¹ в ряді випадків може бути визначена спектроскопічним методом, оскільки часто різні форми А відрізняються своїми спектрами. Наприклад, перетворення о-бензойної кислоти в антрахінон йде через проміжне утворення дегідратованої протонізованої форми о-бензойної кислоти, яка спектром значно відрізняється від вихідної речовини, і, таким чином, концентрація її може бути визначена спектроскопічно.
Між ефективною константою швидкості і константою іонізації існує лінійна залежність, відома під назвою співвідношення Бренстеда – Поляні:
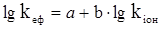 (XII.2.11)
(XII.2.11)
Важливим є також окисно-відновний каталіз. Ключовою стадією багатьох окисно-відновних реакцій є перенесення електрону від одного реагенту до іншого. Це перенесення не завжди відбувається швидко, тоді застосовують каталізатори, що прискорюють стадію перенесення електрона. Каталізаторами цього типу реакцій є комплексні іони перехідних металів з різними ступенями окиснення, наприклад міді, заліза, кобальту, молібдену та ін. Зазвичай, перенесення електрона до реагенту і в зворотному напрямку супроводжується перенесенням атомів і атомних груп, що забезпечують складні хімічні перетворення в каталітичній системі. До реакцій окисно-відновного типу слід віднести розклад пероксиду водню і гідропероксидів органічних сполук, гідрогенування і карбоксилювання, окиснення органічних сполук молекулярним киснем та ін.
Розглянемо розклад пероксиду водню. Перебіг реакції
2Н2О2 → 2Н2О +О2
через циклічний активований комплекс ускладнено внаслідок заборони по орбітальній симетрії і спіну. Тому при відсутності каталізатора вона протікає постадійно за радикально-ланцюговим механізмом. У присутності іонів перехідних металів Cr2O72ˉ, MoO4ˉ, Fe3+, Cu2+ реакція протікає в два етапи. На першому відбувається зв’язування двох молекул пероксиду іоном металу з подальшим перенесенням атомів кисню до металу і виділенням двох молекул води:

Швидкість цієї стадії велика, і в ній встановлюється рівновага. Наявність проміжної сполуки MoO4ˉ з Н2О2 доведено різними фізико-хімічними методами. Друга стадія включає утворення π-звязку між атомами кисню і розпад проміжної сполуки на вихідний каталізатор і молекулярний кисень:

Друга стадія протікає повільно і визначає швидкість всього процесу.
Механізм реакції розпаду Н2О2 формально можна зобразити у вигляді такої схеми:
k1
 1. К + 2Н2О2 [KO2] + 2Н2О
1. К + 2Н2О2 [KO2] + 2Н2О
k2
k3
2. [КO2] К + O2
У присутності каталізаторів швидкість розкладу пероксиду збільшується в 103¸108 раз. Аналогічно протікають реакції гідропероксидів органічних сполук у присутності іонів металів.
Поиск по сайту: