АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомДругоеЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция
Электрокинетические явления
Электрокинетические явления были открыты профессором Ф.Ф. Рейссом в 1908 году при исследовании электролиза воды. Рейсс поставил два эксперимента. В одном из них использовал U-образную трубку, перегороженную диафрагмой из кварцевого песка и заполненную водой (рис. 2.5.3.1).
| Р и с. 2.5.3.3. Схема опыта по возникновению потенциалов течения |
| Р и с. 2.5.3.4. Схема опыта Дорна по возникновению потенциала седиментации |
| h |
| + |
| - |
| Р и с. 2.5.3.1. Схема опыта Рейса по электроосмосу |
| + |
| - |
| Р и с. 2.5.3.2. Схема опыта Рейса по электрофорезу по электрофорезу |
Во втором эксперименте были погружены в глину две стеклянные трубки, заполнены водой и после наложения электрического поля наблюдалось перемещение частиц глины в жидкости в сторону положительно заряженного электрода. Это явление получило название электрофореза (рис. 2.5.3.2). Таким образом, было обнаружено, что частицы имеют заряд, противоположный по знаку заряду жидкости.
Позже было открыто явление, обратное электроосмосу: при течении жидкости через пористое тело под влиянием перепада давлений возникает разность потенциалов. Это явление называется потенциал течения или протекания (рис. 2.5.3.3).
Затем было установлено, что потенциал течения не зависит от размера диафрагмы, количества фильтруемой жидкости, но, как и при электроосмосе, пропорционален объемной скорости фильтрации.
Количественное исследование эффекта, обратного электрофорезу, было проведено Дорном. Он измерял возникающую разность потенциалов при седиментации частиц суспензии кварца в центробежном поле. Явление возникновения разности потенциалов при осаждении дисперсной фазы получило название потенциал седиментации (оседания) или эффект Дорна (рис. 2.5.3.4). Таким образом, по причинно-следственным признакам электрокинетические явления в дисперсных системах делят на 2 группы: в первой движение фаз приводит к возникновению потенциала, во второй - приложение потенциала вызывает движение фаз.
Объяснение этих явлений: на поверхности твердых тел создается двойной электрический слой (ДЭС), имеющий диффузное строение. При относительном перемещении фаз происходит смещение двойного электрического слоя (ДЭС) по плоскости скольжения. Плоскость скольжения обычно проходит по диффузионному слою и часть его ионов остается в дисперсионной среде. В результате дисперсионная среда и дисперсная фаза оказываются заряженными противоположно по знаку. Потенциал, возникающий на плоскости, как уже было сказано ранее, называется электрокинетическим потенциалом (z, дзета). Плоскость скольжения может находиться на разном расстоянии от межфазовой поверхности. Это зависит от скорости движения фаз, вязкости и других факторов. От этих же факторов зависит и значение z-потенциала.
Электроосмос - направленное перемещение жидкости в пористом теле под действием приложенной разности потенциалов (рис. 2.5.3.6).
Капилляр К необходим для точного определения количества вещества, движущейся жидкости, М – мембрана. В прибор заливают раствор и отмечают уровень в капилляре. Если приложить разность потенциалов, то противоионы диффузного слоя, слабо связанные с поверхностью твердого тела, будут перемещаться к соответствующему электроду и благодаря молекулярному трению увлекать за собой дисперсионную среду. Чем меньше потенциал и толщина диффузионного слоя, тем быстрее идет перемещение жидкости в пористом теле. Скорость перемещения жидкости и ее направление (при Е = const) определяется свойствами мембраны и раствора. Количественное изучение позволяет однозначно определить знак z-потенциала и получить зависимость между скоростью перехода жидкости и z-потенциалом. Для получения уравнения электроосмоса необходимо задаться некоторыми ограничениями:
1. Толщина двойного электрического слоя (ДЭС) значительно меньше радиуса пор.
2. Слой жидкости, непосредственно прилегающий к твердой фазе, неподвижен. Движение жидкости в порах ламинарное и подчиняется законам гидродинамики.
3. Распределение зарядов в двойном электрическом слое (ДЭС) не зависит от приложенной разности потенциалов.
4. Твердая фаза - диэлектрик, а жидкость проводит электрический ток.

| Рис. 2.5.3.7. Изменение потенциала j и скорости движения u в капиллярах пористого тела с изменением расстояния от межфазной поверхности |
Для постоянной линейной скорости жидкости относительно мембраны получим выражение:
 .
.
где z - электрическая подвижность;
u 0 - линейная скорость движения жидкости;
h - динамическая вязкость;
e0, e - диэлектрическая проницаемость вакуума и среды;
Е - напряженность электрического поля.
Это классическое выражение для скорости движения жидкости при электроосмосе, оно носит название Гельмгольца-Смолуховского.
Скорость движения дисперсионной среды, отнесенная к единице напряженности электрического поля, называется электроосмотической подвижностью:

Это же уравнение можно написать относительно z-потенциала:

Устойчивость дисперсных систем – это возможность их нахождения в исходном состоянии неопределенно долгое время.
Устойчивость дисперсных систем может быть:
1. К осаждению дисперсной фазы - характеризует способность дисперсной системы сохранять равновесное распределение фазы по объему дисперсионной среды или ее устойчивость к разделению фаз. Это свойство называется седиментационная (кинетическая) устойчивость.
2. К агрегации ее частиц.
Агрегативная устойчивость – это способность дисперсной системы сохранять неизменной во времени степень дисперсности, т.е. размеры частиц и их индивидуальность.
Она обусловлена способностью дисперсных систем образовывать агрегаты (т.е. укрупняться). По отношению к агрегации дисперсные системы могут быть устойчивыми кинетически и термодинамически.
Термодинамически устойчивые системы образуются в результате самопроизвольного диспергирования одной из фаз, т.е. самопроизвольного образования гетерогенной свободнодисперсной системы.
Вспомним, что дисперсные системы также делят на:
· лиофильные, обладающие термодинамической устойчивостью;
· лиофобные, которые термодинамически неустойчивы к агрегации, но могут быть устойчивы кинетически, т.е. обладать значительным временем жизни.
Термодинамическая устойчивость лиофильных систем означает, что они равновесны (энергия Гиббса DG ® min), обратимы и образуются самопроизвольно, как из макрофаз, так и из истинных растворов. Поскольку образуются гетерогенные системы, то поверхностная энергия должна быть скомпенсирована энтропийной составляющей, т.е. частицы дисперсной системы должны участвовать в молекулярно-кинетическом (тепловом) движении. Отсюда следует, что лиофильные системы могут быть только ультромикрогетерогенными.
Лиофобные системы термодинамически неустойчивы, т.к. частицы дисперсной фазы склонны к агрегации. Их агрегативная термодинамическая неустойчивость обусловлена избытком поверхностной энергии. Они не могут быть получены самопроизвольным диспергированием. Для их образования должна быть затрачена внешняя энергия. Укрупнение частиц дисперсной фазы при потере агрегативной устойчивости достигается двумя путями:
1. Изотермическая перегонка, т.е. растворение мелких и рост крупных частиц в соответствии с уравнением Кельвина;
2. За счет слипания частиц, т.е. коагуляцией.
В зависимости от природы среды и концентрации дисперсной фазы эти процессы могут заканчиваться или осаждением, или структурообразованием.
При нарушении агрегативной устойчивости происходит коагуляция.
Коагуляцией называется процесс слипания частиц с образованием крупных агрегатов. В результате коагуляции система теряет свою седиментационную устойчивость, так как частицы становятся слишком крупными и не могут участвовать в броуновском движении.
Коагуляция является самопроизвольным процессом, так как она приводит к уменьшению межфазной поверхности и, следовательно, к уменьшению свободной поверхностной энергии.
Различают две стадии коагуляции.
1 стадия – скрытая коагуляция. На этой стадии частицы укрупняются, но еще не теряют своей седиментационной устойчивости.
2 стадия - явная коагуляция. На этой стадии частицы теряют свою седиментационную устойчивость. Если плотность частиц больше плотности дисперсионной среды, образуется осадок.
Причины коагуляции многообразны. Едва ли существует какое либо внешнее воздействие, которое при достаточной интенсивности не вызывало бы коагуляцию.
1. Минимальная концентрация электролита, при которой начинается коагуляция, называется порогом коагуляции Ck.
2. Коагулирующим действием обладает не весь электролит, а только тот ион, заряд которого совпадает по знаку с зарядом противоиона мицеллы лиофобного золя. Этот ион называют ионом–коагулянтом.
3. Коагулирующая способность иона–коагулянта тем больше, чем больше заряд иона.
Количественно эта закономерность описывается эмпирическим правилом Щульце – Гарди:
 или
или 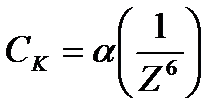 .
.
где a - постоянная для данной системы величина;
Z – заряд иона – коагулянта;
 - порог коагуляции однозарядного, двухзарядного, трехзарядного иона-коагулянта.
- порог коагуляции однозарядного, двухзарядного, трехзарядного иона-коагулянта.
Правило устанавливает, что коагулирующие силы иона тем больше, чем больше его валентность.
Коагулирующая способность иона при одинаковом заряде тем больше, чем больше его кристаллический радиус.
Современная теория устойчивости носит имя ученых Б. В. Дерягина, Л. Д. Ландау, Фервея и Овербека и известна в литературе как теория ДЛФО (DLVO).
Утончение тонкого слоя при сближении частиц происходит путем вытекания из него жидкости. Когда жидкий слой становится достаточно тонким (толщина его менее 100-200 нм), свойства жидкости в нем начинают сильно отличаться от свойств жидкости в окружающем объеме. В слое появляется дополнительное давление, которое Б. В. Дерягин назвал «расклинивающим давлением». По определению Дерягина, оно положительно, когда давление в слое понижено, это противодействует вытеканию из него жидкости, т. е. препятствует сближению коллоидных частиц. Расклинивающее давление может быть и отрицательным, т. е. повышать давление в слое, ускорять вытекание из него жидкости и способствовать сближению частиц.
Возникновение расклинивающего давления в тонких жидких слоях обусловлено, главным образом, двумя факторами:
1) электростатическое взаимодействие в слое - это силы отталкивания с энергией U отт > 0;
2) ван-дер-ваальсовы силы притяжения с энергией U пр < 0.
Результирующая энергия межчастичного взаимодействия U определяется как сумма двух составляющих:

Если  , то преобладают силы отталкивания, коагуляция не происходит, золь является агрегативно устойчивым. В противоположном случае преобладают силы притяжения между частицами, происходит коагуляция.
, то преобладают силы отталкивания, коагуляция не происходит, золь является агрегативно устойчивым. В противоположном случае преобладают силы притяжения между частицами, происходит коагуляция.
Энергия электростатического отталкивания:
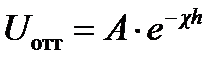 (1)
(1)
где h — расстояние между частицами; 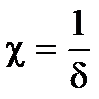 - величина, обратная толщине диффузионного слоя d; А — величина, не зависящая от h и определяемая параметрами ДЭС.
- величина, обратная толщине диффузионного слоя d; А — величина, не зависящая от h и определяемая параметрами ДЭС.
Величины c и А могут быть рассчитаны на основе теории ДЭС.
Энергия притяжения связана, главным образом, с дисперсионным взаимодействием между молекулами. Она может быть рассчитана по уравнению:
 (2)
(2)
где АГ - константа Гамакера. Она рассчитана квантово-статистически и слагается из отдельных констант, характеризующих когезионные и адгезионные взаимодействия,
Результирующая энергия взаимодействия между частицами, находящимися на расстоянии h, определяется уравнением:

 Зависимость суммарной потенциальной энергии межчастичного взаимодействия от расстояния между частицами имеет сложный характер. Общий вид этой зависимости U = f(h) представлен на рис. 1.
Зависимость суммарной потенциальной энергии межчастичного взаимодействия от расстояния между частицами имеет сложный характер. Общий вид этой зависимости U = f(h) представлен на рис. 1.
На графике есть три участка:
1) 0 < h < h1, между частицами преобладают силы притяжения, наблюдается ближний минимум.
U отт ® const; U пр ® -¥.Происходит коагуляция.
2) h 1 < h < h 2 .U(h) > 0 - между частицами преобладают силы отталкивания. Uотт > |Uпр|.
3) h 2 < h < h 3. U(h) < 0 - обнаруживается дальний минимум, однако глубина его невелика.
При h = h 1, h 2и h 3 U(h) = 0, т. е. при этих расстояниях между частицами силы притяжения уравновешиваются силами отталкивания.
Таким образом, если частицы сблизятся на расстояние меньше h 1они неизбежно слипнутся, но для этого должен быть преодолен потенциальный барьер D U k. Это возможно при достаточной кинетической энергии частиц, которая среднестатистически близка к произведению kТ.
Рассмотрим взаимодействие двух частиц. Будем одну частицу считать неподвижной, а вторую - приближающейся к ней с энергией, равной kТ.
Если kТ < D U min, частицы останутся на расстоянии h min и будут связаны между собой через слой дисперсионной среды, т. е. образуют «пару», но непосредственно не слипаются и не теряют своей седиментационной устойчивости. В таких случаях говорят, что взаимодействие происходит в дальнем минимуме.
Если D U min < kТ << D U K, то частицы при столкновении отлетают друг от друга. Система агрегативно устойчива.
Если kТ < D UK, то происходит медленная коагуляция.
Если kТ >DUK,то происходит быстрая коагуляция.
Так как золь обычно рассматривают при постоянной температуре, кинетическая энергия частиц остается постоянной. Следовательно, для коагуляции должен быть уменьшен потенциальный барьер коагуляции D UK.
При коагуляции золя электролитами различают концентрационную и нейтрализационную коагуляцию.
Концентрационная коагуляция имеет место, когда она происходит под действием индифферентного электролита вследствие сжатия диффузного слоя противоионов и уменьшения абсолютного значения z-потенциала.
Нейтрализационная коагуляция происходит при добавлении к золю неиндифферентного электролита. При этом потенциалопределяющие ионы связываются в малорастворимое соединение, что приводит к уменьшению абсолютных величин термодинамического потенциала, а следовательно, и z-потенциала вплоть до нуля.
При коагуляции смесью электролитов различают два типа процессов :
· гомокоагуляция;
· гетерокоагуляция.
Гомокоагуляция - укрупнение подобных частиц в больший агрегат осадка. Причем в процессе отстаивания мелкие частицы растворяются, а крупные увеличиваются за их счет.
Гетерокоагуляция - взаимная коагуляция разнородных дисперсных систем.
При коагуляции золя смесью двух и более электролитов возможны три случая (рис. 3). По оси абсцисс отложена концентрация первого электролита С 1, а C к1 – его порог коагуляции. Аналогично по оси ординат отложена концентрация второго электролита С 2, а С к2 – его порог коагуляции.
1. Аддитивное действие электролитов (линия 1 рис. 3). Электролиты действуют как бы независимо один от другого, их суммарное действие складывается из воздействий каждого из электролитов. Если с1´ - концентрация первого электролита, то для коагуляции золя концентрация второго электролита должна быть равной с2´. Аддитивность наблюдается обычно при сходстве коагулирующей способности обоих электролитов.
2. Синергизм действия (линия 2 рис. 3). Электролиты как бы способствуют друг другу – для коагуляции их требуется меньше, чем нужно по правилу аддитивности (с2″ < c2′). Условия, при которых наблюдается синергизм, сформулировать трудно.
| устойчивое положение |
| z |
| -zкр |
| +zкр |
| х |
| Р и с. 4. Изменение z-потенциала поверхности при ее перезарядке (неправильные ряды) |
Гетероадагуляция - прилипание частиц дисперсной фазы к вводимой в систему чужеродной поверхности.
Одной из причин этого явления является адсорбция стабилизатора на этой поверхности. Например: отложение коллоидных частиц на волокнах при крашении и дроблении.
При введении в коллоидный раствор электролитов, содержащих многовалентные ионы с зарядом противоположные заряду частиц, наблюдается явление «неправильные ряды». Оно состоит в том, что при добавлении к отдельным порциям золя все возрастающего его количества электролита золь сначала остается устойчивым, затем в определенном интервале концентраций происходит коагуляция; далее золь снова становится устойчивым и, наконец, при повышении концентрации электролита опять наступает коагуляция уже окончательная. Подобные явления могут вызывать и большие органические ионы. Объясняется это тем, что при весьма малых количествах введенного электролита ионов недостаточно, чтобы коагулировать золь, т. е. значение x- потенциала остается выше критического (рис. 4). При больших количествах электролита его ионы проявляют коагулирующее действие. Этот интервал концентраций отвечает значениям x- потенциала частиц от x критического первого знака до x критического другого знака.
При еще больших концентрациях многовалентные ионы перезаряжают коллоидную частицу и золь опять устойчивый. В этой зоне x-потенциал опять выше критического значения, но обратен по знаку частицам исходного золя. Наконец, при высоком содержании исходного электролита многовалентные ионы снова снижают значение x-потенциала ниже критического и снова происходит окончательная коагуляция.
Повышение агрегативной устойчивости золя путём введения в него высокомолекулярного соединения (ВМС) называется коллоидной защитой. Происходит образование защитной пленки на поверхности золя (гидратной или ВМС), препятствующей взаимодействию частиц электролита.
Поиск по сайту: