АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомДругоеЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция
XV.6. Електроліз. Напруга розкладу
|
Читайте также: |
Електролізом називають окиснення – відновлення речовин електричним струмом. Ці процеси підкоряються законам Фарадея, про що згадувалось вище. Шляхом електролізу вдається провести процеси, самовільне протікання яких згідно з законами термодинаміки неможливе. Наприклад, розклад 1 моля HCl на елементи супроводжується зростанням енергії Гіббса, рівним 131,26 кДж. Однак, під дією електричного струму цей процес легко може бути здійснений.
При електролізі на негативно зарядженому електроді (катоді) йдуть процеси відновлення, наприклад:
Mz+ + zē → M; Fe3+ + ē → Fe2+; O2 + 4H+ + 4ē → 2H2O
На позитивно зарядженому електроді (аноді) проходять реакції окиснення, характер яких залежить від того, чи здатний розчинятися металевий анод у конкретних умовах електролізу чи він знаходиться в інертному (пасивному) стані. Для розглядуваного анода типова реакція M → Mz+ + zē, для інертного – розряд аніонів і інші окислювальні процеси, наприклад:
4OH- → O2 + 2H2O + 4ē; MnO42-→ MnO4- + ē
До інертних анодів відносяться залізні і нікелеві в лужному середовищі, свинцеві в розчинах, що містять іони SO42-. Високою анодною стійкістю в багатьох середовищах володіє платина.
Процеси, що протікають при електролізі, можна розбити на такі групи:
1. Електроліз, що супроводжується хімічним розкладом електроліта. Наприклад, при електролізі розчину соляної кислоти з використанням інертного аноду йде її розклад:
Катодна реакція 2H+ + 2ēִ → H2
Анодна реакція 2Cl- → Cl2 + 2ē
Загальна реакція в електролізері 2НCl → H2 + Cl2
2. Електроліз, при якому відбувається хімічний розклад розчинника. При електролізі водних розчинів сильних основ (наприклад, NaOH) з інертним анодом на останньому йде реакція:
2ОН- → ½ О2 + Н2О + 2ē
Можна передбачити, що в розчині NaOH можливі такі реакції:
Na+ + ē → Na (ε0=-2,71 B) (1)
2H2O + 2ē → H2 + 2OH- (ε0=-0,82 B) (2)
На катодах з різних металів (окрім ртуті) протікає реакція (2), оскільки їй відповідає більш низький катодний потенціал. Таким чином, в електролізері йде розклад води:
Н2О → Н2 + ½ О2
При електролізі розчинів кисневмісних кислот також відбувається розклад води. Наприклад, при електролізі H2SO4 протікають електродні процеси:
на катоді 2Н+ + 2ē → Н2;
на аноді Н2О → ½ О2 + 2Н+ + 2ē.
Для розряду SO42- з наступним розкладом радикалу SO4, що супроводжується виділенням кисню, вимагається значно більш високий анодний потенціал, ніж для реакції анодного окиснення води.
3. При електролізі розчинів солей ряду металів (Zn, Cd, Cо, Ni, Sn, Pb, Cu, Ag, Au) з розчинними анодами з тих же металів електродні реакції оборотні. Наприклад, при електролізі розчину CuSO4 з мідними електродами на аноді йде реакція Cu → Cu2+ + 2ē, а на катоді Cu2+ +2ē → Cu. Якщо значення анодного і катодного виходів за струмом однакові, то хімічного розкладу електроліту не відбудеться. В процесі електролізу змінюється його вміст біля електродів.
Для здійснення електролізу до ванни необхідно прикласти напругу
Еел = εа – εк + ∑ Е, (ХV. 6.1)
де εа і εк – потенціали аноду і катоду; ∑Е – спад напруги на подоланні омічного опору електроліту, електродів, контактів.
Різниця потенціалів εа і εк поляризованих електродів називається напругою розкладу електроліту. Напруга розкладу не може бути меншою, ніж ЕРС гальванічного елемента, що відповідає оборотній реакції. В табл. 6 наведені Ерозк деяких електролітів.
Таблиця 6
Напруги розкладу Ерозк, (В) деяких електролітів (с =1 моль-екв/л)
| Електроліт | Ерозк | Електроліт | Ерозк | Електроліт | Е розк |
| ZnSO4 | 2,30 | HNO3 | 1,69 | HCl | 1,31 |
| Na2SO4 | 2,21 | NaOH | 1,69 | HBr | 0,94 |
| NaNO3 | 2,15 | H2SO4 | 1,67 | AgNO3 | 0,70 |
| H3PO4 | 1,70 | KOH | 1,67 | HJ | 0,52 |
Ці дані показують, зокрема, що різні електроліти, при електролізі розчинів яких кінцевий результат процесу виявляється однаковим, володіють рівними напругами розкладу. Так, при електролізі водних розчинів H2SO4, HNO3, H3PO4, NaOH, KOH відбувається розклад лише води з виділенням на катоді водню, а на аноді – кисню. Напруга розкладу всіх цих електролітів близька до 1,70 В. Здавалось би, вона повинна відповідати ЕРС ланцюга:
Pt (H2) | кислота | (О2) Pt.
Проте для цього ланцюга ЕРС рівна 1,07 В, а знайдена напруга розкладу більша на 0,63 В. І в інших випадках при електролізі нерідко буває необхідно застосовувати різницю потенціалів більш, ніж ЕРС оборотного ланцюга. Це явище називають перенапругою при електролізі.
Визначають не лише напругу розкладу в цілому, але і відповідні складові її для кожного з електролітів зокрема. Ці складові називають потенціалами виділення або потенціалами розчинення. Потенціал виділення (або розчинення), мабуть, не може бути меншим потенціалу цього електрода при рівноважному процесі в гальванічному елементі. При відсутності побічних процесів він може бути рівним цьому потенціалу, але в більшості випадків він дещо більший за нього. Це явище називають перенапругою на електродах.
Дослідним шляхом встановлено, що потенціали виділення металів (Ag, Zn і ін.), щонайменше при не дуже великих густинах струму, значною мірою рівні або майже рівні їх електродним потенціалам для розчинів даної концентрації, тобто перенапруги для них незначні. Наприклад, потенціал виділення для Cd з розчину CdSO4 (с = 1моль-екв/л) рівний 0,42 В, що співпадає з його рівноважним електродним потенціалом у такому розчині. Однак, для деяких металів при значній швидкості виділення перенапруга досить велика. Так, у заліза при виділенні його з розчину такої ж концентрації при кімнатній температурі вона рівна 0,24 В, для нікелю 0,23 В, для кобальту 0,28 В, але швидко зменшується з підвищенням температури.
Потенціали виділення газів навіть при малих густинах струму можуть бути значно більшими, ніж їх електродні потенціали, а величина перенапруги значно залежить від матеріалу електрода, стану його поверхні та ряду інших факторів.
XV.7 Електрохімічне виділення, анодне розчинення та пасивність металів
Електролітичне виділення металів має широке застосування в металургії для рафінування металів і їх виділення з розчинів, для отримання металевих порошків і гальванічних покрить тощо. Електрохімічні реакції, за виключенням найпростіших реакцій електровідновлення і електроокиснення (утворенням металевої фази або електролітичне розчинення), є надзвичайно складними. Тому і не дивно, що і сьогодні кінетика електроосадження металів вивчена недостатньо. Умови електролізу, що визначають характер кристалізації, мають вирішальний вплив на властивості металу, що виділяється; їх компактність, захисні властивості, міцність щеплення з основним металом, твердість, блиск тощо. В залежності від складу електроліту, густини струму, температури електроліту, характеру його циркуляції можуть утворюватись щільні компактні шари металу (осадки), металеві або блискучі, рихлі губчаті осадки, дендрити, порошкоподібні осадки. При електрохімічному осадженні металів у гідроелектрометалургії і гальванотехніці завдання полягає в одержанні компактних, щільних металевих осадків. Для завдань металокераміки прагнуть одержати порошкоподібні металічні осадки.
Електролітичне виділення металів найчастіше здійснюється з розчинів їхніх простих солей – сульфатів, хлоридів або нітратів. Сумарною катодною реакцією в цьому випадку буде розрядження гідратованих металевих іонів з їх наступним переходом у кристалічну решітку утворюваного на катоді осаду:
Мz+ xH2O + zē = [M] + xH2O
Процес електрокристалізації металів, не дивлячись на свою специфічність, відбувається в основному за законами, які є загальними для процесу утворення кристалів при конденсації пари або виділення твердої фази з розчину. Формування кристалів пов’язане з двома стадіями, що протікають послідовно: утворення кристалічного зародка в середині гомогенної фази (газу, розчину) або на поверхні твердої фази її наступним ростом.
Пересичений пар, пересичений розчин, переохолоджена рідина є метастабільними фазами. Перехід із метастабільного стану в стабільний, що супроводжується зменшенням енергії Гіббса, завжди самочинний процес, за виключенням стадії утворення кристалічних зародків. Зміна енергії Гіббса ΔG, що викликається появою нової фази з радіусом r, рівна:
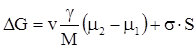 , (XV.7.1)
, (XV.7.1)
де v - об’єм зародку, g – його густина, М- молярна маса, 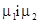 - хімічні потенціали речовини у відповідному середовищі і в новій фазі, σ-поверхневий натяг на межі фаз, S - величина поверхні зародка.
- хімічні потенціали речовини у відповідному середовищі і в новій фазі, σ-поверхневий натяг на межі фаз, S - величина поверхні зародка.
Якщо температура системи вища температури фазового переходу (наприклад, температура плавлення), то μ2 > μ1, і величина ΔG > 0 при будь-яких розмірах зародка. Самовільне утворення зародку в цьому випадку не відбувається. Якщо вихідна фаза пересичена (або переохолоджена), то μ2 < μ1 і ∆G проходить через максимум при деякому критичному розмірі зародка, після якого при подальшому збільшенні його розміру відбувається різке зменшення ∆G. Отже, для утворення нової фази система повинна долати потенціальний бар’єр, який тим вищий, чим більший поверхневий натяг на межі двох фаз.
Зародок нової фази (кристалу чи краплі), що має критичні розміри, знаходиться в стані нестійкої рівноваги. Якщо його розміри дещо зменшаться, то за цим відбудеться подальше самовільне зменшення розміру зародка аж до зникнення. Якщо ж зародок дещо збільшиться, то він буде самовільно рости, оскільки його ріст також веде до зменшення вільної енергії. Радіус рівноважного зародку нової фази r (при його сферичній формі) може бути знайдений за рівнянням Кельвіна:
 , (XV.7.2)
, (XV.7.2)
де c i cs -концентрації пересиченої і насиченої пари, v - молярний об’єм.
При електролізі в залежності від природи металу, складу електроліту, температури ріст кристала з вимірюваною швидкістю вимагає більшого або меншого зміщення потенціалу по відношенню до рівноважного. Рівняння (XV.7.2) в застосуванні до електрокристалізації металу можна записати у вигляді:
 , (XV.7.3)
, (XV.7.3)
де σi -поверхневий натяг її грані, hi -віддаль від точки всередині кристалічного зародку до грані, що задовольняє умові:
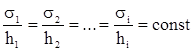 . (XV.7.4)
. (XV.7.4)
Рівності(XV.7.3) визначають залежність між розмірами кристалічного зародка і величиною поляризації h. З (XV.7.3) видно, що поляризація при електрокристалізації пропорційна логарифму степеню пересичення. З чого виходить, що розміри кристалічного зародка тим менші, чим більша поляризація. Роль ефективної концентрації с при електрокристалізації відіграє густина струму.
При електролізі розчинів простих солей характер катодних осадів і величина електродної поляризації визначаються насамперед природою металу, що виділяється (табл.7).
Таблиця 7
Класифікація металів за значенням металевої перенапруги при їх виділенні з розчинів простих солей
| № групи | Метали | Перенапруга, В | Струм обміну, 
| Середні розміри зерен осаду, М |
| І | Hg, Ag, Tl, Pb, Cd, Sn | 
| 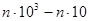
| 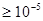
|
| ІІ | Bi, Cu, Zn | 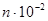
| 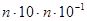
| 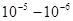
|
| ІІІ | Co, Fe, Ni | 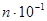
| 
| 
|
Як видно з табл. 7 до першої групи належать метали, що виділяються з водних розчинів або зовсім без перенапруги (ртуть), або з дуже малою перенапругою, яка не перевищує при звичайних густинах струму тисячних часток вольта (Ag, Tl, Pb, Cd, Sn). У цій групі металів (крім ртуті) найбільш чітко виявляється несталість потенціалу в часі, складний характер росту катодного осаду та інші особливості, властиві процесу катодного виділення металів. При промислових густинах струму ці метали дають грубі осади. Струми обміну в металах цієї групи дуже великі. Наприклад, струм обміну між металевою ртуттю і розчином її нітрату перевищує 103  , а між сріблом і розчином нітрату срібла досягає 102 .
, а між сріблом і розчином нітрату срібла досягає 102 .
Бісмут, мідь, цинк становлять другу, проміжну, групу. Для неї характерна металева перенапруга порядку кількох десятків мілівольт, утворення тонших осадів і менші ніж у металів попередньої групи, струми обміну. Для міді, наприклад, струм обміну в контакті з розчином сульфату міді близький до 10-1 .
Найбільшу металеву перенапругу мають метали третьої групи, в яких вона досягає кількох десятих часток вольта. Ці метали виділяються на катоді у вигляді щільних тонкокристалічних осадів. Струми обміну в них малі – для заліза і нікелю в розчинах їхніх сульфатів становлять відповідно 10-4 і 10-5 .
Є група металів – молібден, вольфрам, титан, тантал і ніобій – які взагалі не вдається виділити з водних розчинів у чистому вигляді. Вони виділяються лише у вигляді оксидів, гідроксидів або дуже тонких (до 0,3 мкм) металевих плівок.
Експериментальне дослідження кінетики катодного виділення металів – складне завдання, пов’язане з деякими специфічними особливостями цього процесу. В ході електролізу поверхня катода не стала, а безперервно змінюється внаслідок осадження металу. Характер зростання осаду істотно залежить від природи металу та умов електролізу. Особливо важливо, щоб кристали, що утворюються на поверхні, яка покривається, росли злито і компактно, а не у вигляді одиничних острівців, що, зазвичай, вироджуються в голки або дендрити, або у вигляді хаотичного губчатого утворення кристалів (мікордендритів). При нормальному зростанні осадка відбувається зрощування дрібних груп кристалів, утворюються конгломерати кристалів. Групи кристалів розповсюджуються і вздовж поверхні, зрощуючись вони утворюють суцільний осадок.
Характер осаду і умови його формування в часі при сталій силі струму (або при заданому потенціалі) залежать не лише від природи металу, а й від складу розчину та наявних у ньому домішок. Домішки поверхнево – активних речовин, а також різних окиснювачів (наприклад, розчиненого кисню) впливають на кінетику електровиділення металів. Залежно від ступеня чистоти розчину й природи домішок можуть змінюватись характер росту кристалів, кількість центрів кристалізації, що виникають за одиницю часу на одиниці поверхні катода, значення поляризації при даній густині струму, характер її зміни з часом тощо. У тих випадках, коли катодний вихід металу менший від одиниці (електронегативні метали, високі густини струму), виникають ускладнення, пов’язані зі зміною (звичайно підвищенням) pH прикатодного шару внаслідок видалення водню. Підлужнювання розчину поблизу катода сприяє процесам гідролізу солей металу з утворенням його основних солей і гідроксидів, які можуть впливати на хід електроосадження і включатися в катодний процес.
Катодний процес виділення металів не може протікати без паралельного протікання анодного процесу. Характер протікання анодних процесів відіграє суттєву, а в ряді випадків вирішальну роль, в гідроелектрометалургії, гальваностегії, електросинтезі органічних і неорганічних сполук, корозії металів.
Процеси анодного розчинення металів у більшості випадків є більш складними, ніж простий електрохімічний перехід іона металу з кристалічної решітки в розчин. Для розчинення вимагається не лише сольватація іона, але в ряді випадків також попередня хімічна адсорбція аніонів (Cl-, OH-) із розчину з утворенням перехідного, а потім стійкого комплексу. Для розчинення металу необхідно, щоб його потенціал (потенціал розряду) був більш позитивним, ніж рівноважний ε0. При цьому величина поляризації рівна:
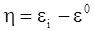 . (XV.7.5)
. (XV.7.5)
Анодне розчинення металу з переходом його в розчин у вигляді простих гідратованих (або у вигляді комплексних) іонів багато в чому є оберненням процесу катодного виділення металів. Анодний процес починається з руйнування кристалічної решітки й закінчується утворенням іонів металу в розчині. Замість стадії формування кристалічної решітки з’являються стадії її руйнування, замість розряду іонів – іонізація атомів металу тощо. Загальну реакцію анодного розчинення металу, якщо утворюються прості гідратовані іони, можна записати у вигляді рівняння:
[M] + хН2O = Mz+ xH2O + zē
або, якщо утворюються комплексні гідратовані іони металу,
[M] + xH2O + yA- = M Ayz-y xH2O + zē.
 Характер анодної поведінки металів залежить від багатьох факторів. Метал, що розчиняється під дією анодної поляризації, при зміні умов може втратити цю здатність і перетворитися в нерозчинний анод. Таке перетворення розчиненого анода в нерозчинний є окремим випадком пасивності металів.
Характер анодної поведінки металів залежить від багатьох факторів. Метал, що розчиняється під дією анодної поляризації, при зміні умов може втратити цю здатність і перетворитися в нерозчинний анод. Таке перетворення розчиненого анода в нерозчинний є окремим випадком пасивності металів.
Переведення металу в пасивний стан досягається не лише при дії відповідних окиснювачів (прикладом чого є пасивація заліза концентрованим розчином азотної кислоти), а й іншими способами, зокрема анодною поляризацією. Найвиразніше це явище виявляється на потенціостатичних кривих потенціал анода – густина струму і-ε (рис.88). Спочатку зі зростанням анодного потенціалу зростає густина анодного струму (ділянка АВ) і швидкість розчинення металу. Таку залежність можна спостерігати до тих пір, доки не буде досягнутий потенціал утворення оксиду, наприклад, за реакцією:
хM + 2уOH- → MхOу + уH2O + 2уē
Плівка оксидів, що утворюється створює перешкоду для переходу іонів металу в розчин. Швидкість розчинення металу різко падає (відрізок ВС), виникає пасивація металу. Далі відмічається область стійкої пасивації, що характеризується незалежністю густини струму від потенціалу (відрізок СД). Ділянка потенціостатичної поляризаційної кривої, що сполучає область активного стану з областю пасивності, називається пасивною областю. Нарешті, при деякому значенні потенціалу (точка Д) відбувається підвищення густини струму, що пов’язане зі збільшенням швидкості розчинення. Ця зона потенціалів називається областю перепасивації, або транспасивації. Для неї характерне розчинення металу у вигляді іонів вищої валентності, ніж при його розчиненні в активному стані. Отже при зміні потенціалу в бік більш позитивних значень метал послідовно проходить через активний, перехідний (або перед- пасивний), пасивний і транспасивний стани. Зміщуючи потенціал від позитивних значень до негативних, можна примусити метал пройти через ті самі стани, але в зворотному порядку. Для характеристики потенціостатичної кривої найважливішими є точки переходу з одного стану в інший. Потенціал, що відповідає точці В (рис. 88), при якому починається перехід металу з активного стану в пасивний, називається потенціалом початку пасивації Eп, або потенціалом пасивації.
При потенціалі, що відповідає точці С, метал уже пасивний. Проте навіть його незначне відхилення в негативний бік порушує його пасивний стан, і метал починає активуватися. Цей потенціал називають потенціалом активації Еакт, або Фладе – потенціалом (за ім’ям німецького вченого), причому останній термін часто вживають і для потенціалу пасивації Еп.
Наступним характеристичним потенціалом є потенціал, що відповідає точці Д і називається потенціалом депасивації Ед, при якому метал в області пасивного стану вступає в область транспасивності. Для кожної з областей потенціостатичної кривої, розмежованих критичними точками, характерний свій закон зміни швидкості розчинення (зміни анодної густини струму) з потенціалом.
Зазначимо, що область перепасивації, тобто перехід металу в новий активний стан і підвищення швидкості його розчинення при досягненні достатньо позитивних потенціалів, спостерігається не завжди. Проте і в цих випадках після досягнення певного значення потенціалу виявляється збільшення густини струму. Але тут він відповідає не поновленню розчинення металу, а початку виділення кисню. Відомі також випадки, коли після перепасивації настає друга область пасивності, яка може потім змінитися новим збільшенням струму, пов’язане з розчиненням металу (але вже у вигляді інших іонів) або з виділенням кисню.
Описана картина пасивації може набути й іншого вигляду в залежності від хімічних, електрохімічних і механічних властивостей плівки, що утворюється, і від особливостей її взаємодії з розчином. Згідно з плівковою теорією пасивність пов’язана з утворенням на поверхні металу окремої фази –фазо-вираженого оксиду або іншої сполуки.
Перша стадія утворення оксидного шару – адсорбція (хемосорбція) кисню. На платині процес на цій стадії припиняється, і на її поверхні знаходиться, в залежності від умов, незаповнений або заповнений моношар адсорбованого кисню. На інших металах утворення шару продовжується. Після того, як товщина досягає 2–3 атомних розмірів, шар перетворюється в окрему поверхневу фазу кристалічної (рідше аморфної) будови із властивостями відповідних об’ємних оксидів.
У залежності від структури металу і оксиду поверхневий фазовий шар має різнобічний характер. На деяких (цинк, кадмій, магній і ін.) утворюються щільні мало пористі або безпористі шари, товщина яких не більше 1 мкм. У ряді випадків (наприклад, на залізі) утворюються суцільні плівки, в яких під впливом структури металу – підкладки, а також через дію поверхневих хімічних сил кристалічна решітка викривлена. Фізико – хімічні і термодинамічні параметри таких плівок відрізняються від параметрів звичайних об’ємних оксидів. Через наявність внутрішніх напруг деформованої решітки такі плівки стійкі лише при незначній товщині, приблизно до 3 – 5 нм.
На поверхні металевих електродів, як правило, одночасно утворюються різі види оксидних шарів, наприклад, пористі фазові шари над адсорбційними шарами. Часто відбувається процес старіння оксидних шарів – зміна їх властивостей у часі або перехід із одного виду в другий.
Поведінка металевих електродів з окисненою поверхнею залежить від властивостей оксидних шарів. Навіть незначна кількість хемосорбованого кисню різко міняє будову подвійного електричного шару; а також впливає на адсорбцію інших речовин. При проходженні струму пористі шари екранують значну частку ділянок поверхні і гальмують підведення до них реагентів і відведення продуктів реакції; окрім того, через підвищену густину струму в порах збільшується омічне падіння напруги. Все це утруднює протікання електрохімічних реакцій, зокрема, реакцій подальшого зростання товщини шару.
Для електродів з безпористими або дуже малопористими шарами з чисто іонною провідністю електрохімічна реакція можлива лише на внутрішній (на межі з металом) поверхні шару.
Якщо поряд з іонною в шарі є і електронна провідність, то електрохімічна реакція частково або повністю витісняється на внутрішню поверхню шару. Крім того, на цій поверхні можливе протікання й інших електрохімічних реакцій з участю компонентів розчину, а зокрема, анодного виділення кисню.
В окремих випадках при анодній поляризації металів у розчинах електроліту утворюються поверхневі шари – адсорбційні або фазові, що містять не кисень, а аніони з розчину. Так, при анодній поляризації срібла в розчині, що містить хлорид – іони утворюється поверхневий шар хлориду срібла, при анодній поляризації свинцю в розчині сульфатної кислоти – шар сульфату свинцю. Таким же шляхом утворюються шари із сульфідів, фосфатів і інших солей. Властивості таких сольових шарів багато де в чому аналогічні властивостям оксидних шарів.
Поиск по сайту: