АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомДругоеЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция
ВИДЫ ПОВРЕЖДЕНИЙ И ГИБЕЛИ КЛЕТОК. УНИВЕРСАЛЬНЫЙ ОТВЕТ КЛЕТКИ НА ПОВРЕЖДЕНИЕ
Повреждение клетки - типический патологический процесс, основу которого составляют нарушения внутриклеточного гомеостаза, приводящие к нарушению структурной целостности клетки и ее функциональных способностей после удаления повреждающего агента. Так, например, на первом этапе нарушение функционирования клетки, вызванное действием неблагоприятных факторов, например недостатком кислорода или действием токсических соединений, может и не привести к повреждению клетки: как только восстановятся нормальные окружающие условия, клетка вновь вернется в состояние, близкое к исходному. Например, если в каком-нибудь участке миокарда кровоснабжение прекращается на короткий промежуток времени (не более 10-15 мин), а затем восстанавливается, то кардиомиоциты сохраняют способность к регенерации и нормальному функционированию. Если кровоснабжение не восстанавливается, то повреждение миокарда становится необратимым и кардиомиоциты на этом участке погибают.
Различают непосредственное (первичное) и опосредованное (вторичное) повреждения. Последнее возникает как следствие первичных нарушений постоянства внутренней среды организма.
В зависимости от скорости развития и выраженности основных проявлений повреждение клетки может быть острым и хроническим.
Острое повреждение развивается быстро, как правило, в результате однократного, но интенсивного повреждающего воздействия, в то время как хроническое повреждение протекает медленно и является следствием многократных, но менее интенсивных патогенных влияний.
В зависимости от периода жизненного цикла, на который приходится действие повреждающего агента, повреждение клетки может быть митотическим и интерфазным.
В зависимости от степени нарушения внутриклеточного гомеостаза повреждение бывает обратимым и необратимым (см. выше).
Выделяют два патогенетических варианта повреждения клеток:
1. Насильственный. Развивается в случае действия на исходно здоровую клетку физических, химических и биологических факторов, интенсивность которых превышает обычные возмущающие воздействия, к которым клетка адаптирована. Наиболее чувствительны к данному варианту повреждения функционально малоактивные клетки, обладающие малой мощностью собственных гомеостатических механизмов.
2. Цитопатический. Возникает в результате первичного нарушения защитно-компенсаторных гомеостатических механизмов клетки. В этом случае фактором, запускающим патогенетические механизмы повреждения, являются естественные для данной клетки возмущающие стимулы, которые в этих условиях становятся повреждающими. К цитопатическому варианту относятся все виды повреждения клетки, возникающего вследствие отсутствия какихлибо необходимых ей компонентов (гипоксическое, нервнотрофическое, при голодании, гиповитаминозах, недостаточности антиоксидантной системы, генетических дефектах и др.). К цитопатическому повреждению наиболее чувствительны те клетки, реактивность, а следовательно, и функциональная активность которых в естественных условиях очень высоки (нейроны, кардиомиоциты).
• Причинами повреждения клеток могут быть следующие факторы: гипоксия. Чрезвычайно важная и распространенная причина повреждения клеток. Уменьшение кровообращения (ишемия), возникающее при атеросклерозе, тромбозе, сдавлении артерий, является основной причиной гипоксии. Другой причиной может быть недостаточная оксигенация крови при сердечно-сосудистой или легочной патологии. Третьей причиной может являться нарушение транспорта кислорода, например при анемии, отравлении окисью углерода или действии метгемоглобинобразователей (нитраты и нитриты, хлорноватые и хлорноватистые соли, феррицианиды, лекарственные вещества - фенацетин, амидопирин, сульфаниламиды и др.) (подробнее см. раздел 16.2);
• физические агенты - механическая травма, температурные воздействия, колебания барометрического давления, ионизирующая и ультрафиолетовая радиация, электрический ток;
• химические агенты и лекарства. Повреждение клеток может быть вызвано как жизненно необходимыми химическими соединениями, такими, как, например, глюкоза или поваренная соль в гипертонических концентрациях, кислород в высоких концентрациях. Вещества, известные как яды (в частности, мышьяк, цианиды, соли ртути), могут вызывать гибель клеток в считанные минуты или часы. Гибель клеток может наступать при действии факторов внешней среды, «социальных» факторов - алкоголя, курения, наркотиков и др.;
• иммунологические реакции. Хотя иммунные реакции защищают организм от воздействия биологических агентов, в ряде случаев (аллергия, аутоиммунные реакции) они могут обусловливать повреждение клеток;
• генетические повреждения (например, наследственные мембранопатии, энзимопатии и др.);
• дисбаланс питания.
Первое событие, которое в конце концов приводит к повреждению клетки, - это взаимодействие повреждающего агента с мишенями-молекулами (табл. 3-1). Так, мишенями для ультрафиолетовых лучей могут быть ароматические группы белков, ферментов и рецепторов или нуклеотиды в молекулах ДНК и РНК. Мишенью для окиси углерода служат различные гемсодержащие ферменты. Мишенью при действии гипоксии оказываются митохондрии, которые перестают запасать энергию в форме АТФ, и т.д.
Таблица 3-1. Примеры повреждающих агентов, действующих на клетку
| Действующие агенты | Основные мишени | Первичные процессы |
| Токсины | Активные центры ферментов и рецепторов, ионные каналы | Инактивация ферментов, блокада рецепторов и ионных каналов |
| Ультрафиолетовое излучение | Нуклеиновые кислоты и белки | Фотохимические реакции нуклеотидов и определенных аминокислот |
Окончание табл. 3-1
| СВЧ миллиметрового диапазона | Молекулы воды | Ускорение процессов, лимитируемых диффузией в водной среде |
| Гипоксия Митохондрии Снижение синтеза АТФ | ||
| Гиперкалиемия | Клеточные мембраны | Увеличение мембранного потенциала*, гиперполяризация |
* Увеличение разницы потенциалов между наружной и внутренней поверхностью клеточной мембраны.
Взаимодействие повреждающего фактора c мишенью может приводить к повреждению самой мишени, что наблюдается, например, при действии ультрафиолетовых лучей на клетки. В других случаях мишень не повреждается действующим агентом, но временно перестает функционировать. Именно это приводит в конечном счете к повреждению клетки в целом. Например, при действии цианистого калия выключается функция цитохромоксидазы, которая в данном случае служит мишенью для яда. Но фермент не повреждается: если удалить цианид из окружающей среды, функция цитохромоксидазы восстановится. Причиной гибели клетки является последующее повреждение клеточных структур, вызванное длительным прекращением энергообеспечения.
Таким образом, между моментом взаимодействия повреждающего агента с мишенью и процессом повреждения определенных клеточных структур может произойти целая цепь последовательных событий.
Гибель клетки - это конечный результат ее повреждения. Существует два основных типа клеточной гибели - некроз и апоптоз. На сегодняшний день выделяют также третий тип смерти клеток - конечное дифференцирование, который, по мнению большинства современных ученых, является одной из форм апоптоза.
Некроз (от греч. nekros - мертвый) - это патологическая форма гибели клетки вследствие ее необратимого химического или физического повреждения (высокая и низкая температура, органические растворители, гипоксия, отравление, гипотонический шок, ионизирующее излучение и др.). Некроз представляет собой спектр морфологических изменений, являющихся результатом разрушающего действия ферментов на поврежденную клетку. Развивается два конкурирующих процесса: ферментативное переваривание клетки
(колликвационный, разжижающий некроз) и денатурация белков (коагуляционный некроз). Для проявления обоих этих процессов требуется несколько часов, поэтому в случае внезапной смерти, например, при инфаркте миокарда соответствующие морфологические изменения просто не успевают развиться. Этот вид гибели клеток генетически не контролируется.
Некрозу могут предшествовать периоды паранекроза и некробиоза.
Паранекроз - заметные, но обратимые изменения в клетке: помутнение цитоплазмы, вакуолизация, появление грубодисперсных осадков, увеличение проникновения в клетку различных красителей.
Некробиоз -состояние «между жизнью и смертью» (от necros - мертвый и bios - живой); изменения в клетке, предшествующие ее смерти. При некробиозе в отличие от некроза возможно возвращение клетки в исходное состояние после устранения причины, вызвавшей некробиоз.
Если некроз считается патологической формой клеточной гибели, возникающей в результате чрезмерного (резкого, сильного) повреждающего воздействия на клетку, то апоптоз противопоставляется ему как контролируемый процесс самоуничтожения клетки.
Апоптоз (от греч. аро - отделение и ptosis - падение) - это генетически контролируемая физиологическая форма гибели клетки. Биологическое значение апоптоза заключается в поддержании внутреннего гомеостаза организма на клеточном, тканевом и системном уровнях. Апоптоз ответствен за многочисленные физиологические и патологические процессы в организме:
1. Программированное разрушение клеток на стадии эмбриогенеза (автономный апоптоз). Различают три категории автономного апоптоза: морфогенетический, гистогенетический и филогенетический.
Морфогенетический апоптоз участвует в разрушении различных тканевых зачатков, что обеспечивается:
• гибелью клеток в межпальцевых промежутках;
• гибелью клеток «избыточного» эпителия при слиянии нёбных отростков, когда формируется твердое нёбо;
• гибелью клеток в дорсальной части нервной трубки во время смыкания, что необходимо для достижения единства эпителия двух сторон нервной трубки и связанной с ними мезодермы.
Нарушение морфогенетического апоптоза в этих трех локализациях приводит, соответственно, к развитию синдактилии, расщеплению твердого нёба и spina bifida.
Гистогенетический апоптоз имеет место при дифференцировке тканей и органов, например, при гормонально-зависимой дифференцировке половых органов из тканевых зачатков. Так, клетками Сертоли в яичках плода мужского пола синтезируется гормон, который вызывает путем апоптоза регрессию протоков Мюллера, из которых у женщин формируются маточные трубы, матка и верхняя часть влагалища.
Филогенетический апоптоз участвует в удалении рудиментарных структур у эмбриона, например пронефроса.
2. Гормонозависимая инволюция органов у взрослых, например отторжение клеток эндометрия во время менструального цикла, атрезия фолликулов в яичниках в менопаузе, регрессия молочной железы после прекращения лактации.
3. Стабилизация численности клеток и их популяций в активно пролиферирующих тканях, например клеток эпителия кишечника, крови и иммунной системы; удаление стареющих клеток, прошедших свой жизненный цикл.
4. Элиминация части опухолевых клеток во время спонтанной регрессии опухолей.
5. Гибель клеток иммунной системы (В- и Т-лимфоцитов) при гипосекреции цитокинов, аутореактивных Т-клеток в тимусе - при их клональной делеции.
6. Патологическая атрофия гормонозависимых органов, например атрофия предстательной железы после кастрации; истощение лимфоцитов в тимусе на фоне терапии глюкокортикоидами.
7. Патологическая атрофия паренхиматозных органов после обтурации выводящих протоков, например, в поджелудочной и слюнных железах, почках.
8. Гибель клеток, вызванная действием цитотоксических Т-лимфоцитов, в частности при отторжении трансплантата и болезни «трансплантат против хозяина».
9. Элиминация клеток, инфицированных вирусами (например, при вирусном гепатите фрагменты апоптотических клеток обнаруживаются в печени в виде телец Каунсильмана).
10. Элиминация поврежденных клеток при действии химических и физических факторов (высокая и низкая температура,
ионизирующее излучение, противоопухолевые препараты и др.) в дозе, недостаточной для развития некроза.
Апоптоз является активным процессом саморазрушения клетки, по морфологическим и другим признакам он существенно отличается от некроза (см. табл. 3-2). Наиболее характерные проявления апоптоза определяются тем, что первые события, связанные с его осуществлением, начинаются в ядре. К ним относятся конденсация хроматина с формированием скоплений (в виде ленты, комочков), прилежащих к ядерной мембране (маргинация хроматина), и появление вдавлений ядерной мембраны, приводящих к фрагментации ядра (кариорексису) и образованию апоптотических телец - внеклеточных фрагментов ядра, окруженных мембраной. В цитоплазме происходит конденсация и сморщивание гранул. Клеточная мембрана утрачивает ворсинчатость, образует пузыревидные вздутия, на ней экспрессируются различные молекулы, распознаваемые фагоцитами (фосфатидилсерин, тромбоспондин, десиалированные мембранные гликоконъюгаты). От поверхности апоптотической клетки отщепляются небольшие везикулы, наполненные содержимым цитоплазмы (митохондрии, рибосомы и др.) и окруженные мембранным липидным бислоем. Клетка постепенно уменьшается в объеме, округляется и теряет межклеточные контакты. Апоптотические клетки и их фрагменты (апоптотические тельца, везикулы) поглощаются макрофагами, нейтрофилами и другими соседними клетками, не являющимися «профессиональными» фагоцитами. В результате эндоцитоза содержимое апоптотических клеток не выделяется в межклеточное пространство, как это происходит при некрозе, при котором вокруг гибнущих клеток скапливаются их активные внутриклеточные компоненты, включая энзимы, закисляется среда, что способствует повреждению соседних клеток и развитию воспалительной реакции, т.е. апоптоз одиночной клетки не отражается на ее окружении.
В развитии апоптоза выделяют 3 стадии: сигнальную (индукторную), эффекторную и деградации (деструкции).
Пусковыми факторами апоптоза могут быть как внешние (внеклеточные) факторы, так и внутриклеточные сигналы. Сигнал воспринимается клеткой, далее последовательно передается молекулам-посредникам (мессенджерам) различного порядка и достигает ядра, где происходит включение программы клеточного «самоубийства».
Таблица 3-2. Дифференциальные признаки некроза и апоптоза
| Признаки Некроз Апоптоз | ||
| Пусковой фактор | Разрушение мембраны под действием патологических стимулов | Деградация ДНК под действием физиологических и патологических стимулов |
| Распространенность Группа клеток Одиночная клетка | ||
| Биохимические изменения в клетке | Активация лизосомальных ферментов | Активация эндонуклеаз, фрагментирующих ДНК |
| Энергозависимость Нет Есть | ||
| Целостность цитоплазматической и внутриклеточных мембран | Нарушена | Сохранена |
| Морфологические изменения клетки | Увеличение размеров клетки, разрыхление мембраны, набухание (окноз) цитоплазмы, митохондрий, лизис ядра и гранул | Уменьшение размеров клетки, уплотнение и вздутие мембраны, кариопикноз, кариорексис, маргинация хроматина, конденсация и уплотнение гранул |
| Воспалительный ответ | Есть | Нет |
| Элиминация гибнущей клетки | Лизис клетки, фагоцитоз | Фрагментация клетки, поглощение фрагментов клетки (мембранных везикул, апоптотических телец) соседними клетками и фагоцитами |
Классическими индукторами экзогенного апоптоза являются стероидные гормоны (половые, тиреоидные, кальцитриол, минералокортикоиды, ретиноиды), антигены, антитела, митогены, цитокины (фактор некроза опухолей (TNF) α, интерлейкин (IL) 1, IL-10, интерферон (INF) γ, β-токоферол и др.). Их проапоптогенное действие осуществляется через ядерные рецепторы (например, GR - глюкокортикоидный рецептор), специализированные мембранные «рецепторы смерти» (Fas, TNF-RI, TNF-RII, DR-3, DR-5 и др.) и рецепторы, выполняющие иные функции, например функцию активации клетки (T-клеточный рецептор (TCR),
цитокиновые рецепторы), что сопровождается развитием активационного апоптоза.
Ситуация эндогенного запуска программы гибели клетки возможна при лишении ее ростовых факторов (IL-2, IL-3, IL-4, INF-α, колониестимулирующих факторов - гранулоцитарно-макрофагального (ГМ-КСФ), гранулоцитарного (Г-КСФ), эритропоэтина и др.), нарушении контактов с внеклеточным матриксом и другими клетками, накоплении нерепарируемых разрывов ДНК (например, при повреждении клетки вирусами, ионизирующей радиацией, ультрафиолетовым излучением, токсинами и др.). В последнем случае важная роль отводится ядерному белку р53 (см. ниже).
В результате запуска апоптогенным (экзогенным или эндогенным) сигналом программы активации генов-индукторов апоптоза (Р53, BAX, PIG, FAS/APO-1, IGF-BP3 и др.) и/или угнетения апоптозингибирующих генов (генов семейства BCL-2) в клетке изменяется набор внутриклеточных РНК и белков, синтезируются и активируются ферменты, способные разрушать клеточные белки (протеазы - каспазы, катепсины, кальпаины, гранзимы) и нуклеиновые кислоты (нуклеазы - Са2+/Мg2+-зависимая эндонуклеаза и др.). Основным проявлением деструктивных изменений клетки при апоптозе является деградация хроматина, основой которого служит расщепление ДНК.
В настоящее время выделены несколько основных механизмов реализации апоптоза:
1) Рецепторный. Осуществляется с помощью «рецепторов смерти» (см. выше) при активирующем взаимодействии с соответствующими лигандами, большинство из которых относится к суперсемейству фактора некроза опухолей. Взаимодействие рецептора с лигандом приводит к активации адапторных белков, ассоциированных с «доменами смерти» (FADD - Fas-associated death domain, TRADD - TNF-R-associated death domain), и прокаспазы 8, продукт которой - каспаза 8 (инициаторная) активирует каспазу 3 (эффекторную), что, в свою очередь, обусловливает активацию эндонуклеаз, фрагментирующих ДНК.
2) Митохондриальный. Участие митохондрий в апоптозе обеспечивается присутствием в их матриксе и межмембранном пространстве большого количества биологически активных веществ (цитохрома С (Cyt С); прокаспаз 2, 3, 9; апоптозиндуцирующего фактора (AIF), обладающих выраженным апоптогенным действием. Фактором активации апоптоза является выход данных веществ
в цитоплазму при снижении трансмембранного потенциала митохондрий вследствие открытия гигантских митохондриальных пор (выполняют роль Ca2+-, рН-, потенциал-, НАДФ2Н/НАДФ+- и редоксзависимых каналов) и повышения проницаемости митохондриальных мембран. К раскрытию пор приводят истощение в клетках восстановленного глутатиона, НАДФН, АТФ и АДФ, образование активных форм кислорода, разобщение окислительного фосфорилирования, увеличение содержания Ca2+ в цитоплазме. Поступление межмембранных белков и активация апоптоза возможны также при разрыве наружной мембраны митохондрий вследствие гиперполяризации внутренней мембраны.
3) р53-опосредованный. p53 - многофункциональный белок, играющий важную роль в мониторинге сигналов о состоянии клетки, целостности ее генома, активности систем ДНК-репарации. Повреждение ДНК ведет к накоплению белка р53 в клетке. Это определяет остановку клеточного цикла в фазах G1 и G2, предотвращает репликацию, активирует синтез и репарацию ДНК, а следовательно, создает условия для восстановления нативной структуры ДНК, препятствует появлению мутантных и анеуплоидных клеток в организме. В случае если имеется недостаточность систем ДНК-репарации и повреждения ДНК сохраняются, клетка подвергается апоптозу. В частности, белок р53 способен индуцировать транскрипцию таких апоптогенных факторов, как Bax, Fas- рецептор, DR-5 и др.
4) Перфорин-гранзимовый. Цитотоксические Т-лимфоциты (Т-киллеры) вызывают апоптоз клеток-мишеней (например, инфицированных клеток) с помощью белка перфорина. Полимеризуясь, перфорин образует в цитоплазматической мембране клеткимишени трансмембранные каналы, по которым внутрь клетки поступают секретируемые Т-киллером гранзимы (фрагментины) - смесь сериновых протеаз. Основным компонентом этой смеси является гранзим В - протеолитический фермент, активирующий каспазу 3.
Важную роль в процессе передачи апоптогенного сигнала и регуляции апоптоза играют следующие внутриклеточные факторы (мессенджеры):
• концентрация ионов Ca (Ca2+ активирует сериновые и цистеиновые протеазы, Ca2+/Mg2+-зависимую эндонуклеазу);
• протеинкиназы А (медиатор апоптоза) и С (ингибитор апоптоза);
• церамид (стимулирует киназы, фосфатазы);
• активные формы кислорода (обусловливают снижение трансмембранного потенциала митохондрий, увеличение внутриклеточной концентрации Ca2+, образование цАМФ);
• монооксид азота (опосредует изменение экспрессии р53, открытие гигантских пор в митохондриях и снижение митохондриального потенциала).
При различных патологических процессах в организме (инфекция, воспаление, иммунодефициты, гипо- и апластическая анемии, опухоли и др.) могут наблюдаться как ускорение, так и замедление апоптоза.
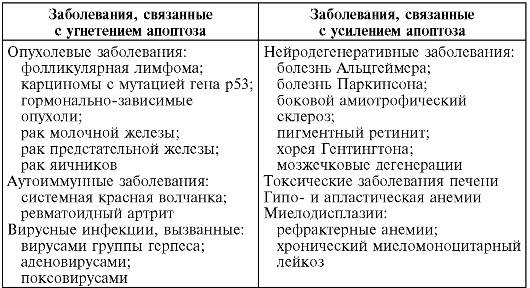
Примеры некоторых заболеваний, в патогенез которых включается апоптоз, представлены в табл. 3-3.
Таблица 3-3. Примеры заболеваний, связанных с угнетением или усилением апоптоза
 Универсальный ответ клетки на повреждение. Особенностью развития патологических изменений в клетках в ответ на самые различные неблагоприятные воздействия является сходство этих изменений, которое позволило Д.Н. Насонову и В.Я. Александрову выдвинуть в 1940 г. теорию о неспецифической реакции клеток на повреждение. Ее суть сводится к следующему - каким бы ни был повреждающий агент и на какие бы клетки он ни действовал, ответ клеток по ряду показателей является одинаковым. К числу таких показателей относятся:
Универсальный ответ клетки на повреждение. Особенностью развития патологических изменений в клетках в ответ на самые различные неблагоприятные воздействия является сходство этих изменений, которое позволило Д.Н. Насонову и В.Я. Александрову выдвинуть в 1940 г. теорию о неспецифической реакции клеток на повреждение. Ее суть сводится к следующему - каким бы ни был повреждающий агент и на какие бы клетки он ни действовал, ответ клеток по ряду показателей является одинаковым. К числу таких показателей относятся:
1) уменьшение дисперсности коллоидов цитоплазмы и ядра;
2) увеличение вязкости цитоплазмы, которому иногда предшествует ее некоторое уменьшение;
3) увеличение сродства цитоплазмы и ядра к ряду красителей. Во многих случаях обнаруживаются также набухание клетки,
изменение ионной проницаемости плазматической и внутриклеточных мембран, выход метаболитов из клетки, изменение флуоресценции, повышение кислотности цитоплазмы и т.д. Существование такого стереотипа изменений физико-химических свойств клеток при их повреждении связано с тем, что молекулярноклеточные механизмы повреждения сходны, хотя причины, вызвавшие повреждение, могут быть самыми разными. Практически у всех клеток при действии повреждающих агентов наблюдается резкое увеличение проницаемости клеточных мембран для ионов кальция. Это сопровождается активацией различных внутриклеточных ферментов и процессов: протеинкиназ, фосфолипаз, фосфодиэстеразы циклических нуклеотидов, системы биосинтеза белков и т.д. Эти изменения могут быть обратимыми, но в конце концов при сильном и длительном воздействии повреждающего фактора происходит стойкое нарушение функций клеток, а следовательно, ткани и органа в целом.
Поиск по сайту: