АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомДругоеЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция
ХІ.17. Гетерогенні процеси
Гетерогенними називають процеси, що відбуваються на поверхні розділу фаз, які дотикаються. Існують три типи гетерогенних процесів:
1) які протікають на поверхні розділу фаз, що взаємодіють − розчинення солей, металів у кислотах; поглинання оксиду вуглецю (ІV) твердим лугом тощо. Схематично вони можуть бути позначені як “тв+р”, “тв+газ”;
2) протікають лише в об’ємі однієї з фаз внаслідок дифузії до неї реагуючих компонентів з іншої фази. Сюди відносять реакції “р+р”, окиснення металів, окиснення в розчинах;
3) які протікають на поверхні твердої фази, що утворюється (або на поверхні розділу нової і вихідної фаз) − реакції двох твердих тіл, перетворення типу “тв+газ=тв” (відновлення-окиснення), поліморфні перетворення (Snбіле → Snсіре) − твердофазні перетворення.
Швидкість гетерогенних процесів залежить від розмірів і стану поверхні розділу фаз, а також від швидкості їх відносного руху. Як випливає з вищесказаного, гетерогенні процеси багатостадійні. Окрім основного процесу, що протікає на поверхні розділу фаз, обов’язковими є стадії підведення до цієї поверхні вихідних речовин і відведення від неї продуктів реакції. Внаслідок того, що ці стадії протікають послідовно, швидкість сумарного процесу визначається найбільш повільною стадією. Якщо визначаючою стадією є хімічна реакція на поверхні розділу фаз, то гетерогенний процес описується законами хімічної кінетики і, значить, протікає в кінетичній області. Якщо, як це часто буває, найбільш повільно здійснюється підведення і відведення відповідних речовин, то гетерогенний процес описується законами дифузії, тобто він протікає в дифузійній області (рис. 66). Температура сильніше впливає на швидкість хімічних процесів, ніж на дифузію, тому гетерогенна хімічна реакція при підвищенні температури може перейти із кінетичної області в дифузійну.
 Дифузія має велике значення в гетерогенних процесах, оскільки за рахунок її відбувається зміна концентрації в приповерхневому шарі, що впливає на кінетику процесу. Дифузія описується законами Фіка.
Дифузія має велике значення в гетерогенних процесах, оскільки за рахунок її відбувається зміна концентрації в приповерхневому шарі, що впливає на кінетику процесу. Дифузія описується законами Фіка.
Перший закон Фіка стверджує, що маса речовини dm, яка переноситься шляхом дифузії в напрямку вісі х через перпендикулярну цьому напрямку площину, пропорційна площі S цієї площини, часу dτ і градієнту концентрації 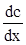 вздовж вибраного напрямку:
вздовж вибраного напрямку:
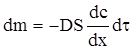 , (ХІ.17.1)
, (ХІ.17.1)
де D − коефіцієнт дифузії. Знак мінус у цій формулі показує, що процес дифузії спрямований у бік зменшення концентрації.
На підставі цього рівняння швидкість дифузії можна записати таким чином
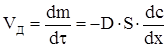 . (ХІ.17.2)
. (ХІ.17.2)
Коефіцієнт дифузії залежить від температури: для рідких і газуватих середовищ він збільшується з ростом температури на 10° приблизно на 20%.
Швидкість дифузії збільшується з підвищенням температури за законом, аналогічному рівнянню Арреніуса: 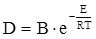 . Проте величина енергії активації дифузійного процесу Е, зазвичай, не перевищує 5 − 20 кДж/моль, тобто вона значно менша енергії активації більшості хімічних реакцій. Тому зміна температури набагато слабше впливає на швидкість дифузійних процесів, ніж хімічних.
. Проте величина енергії активації дифузійного процесу Е, зазвичай, не перевищує 5 − 20 кДж/моль, тобто вона значно менша енергії активації більшості хімічних реакцій. Тому зміна температури набагато слабше впливає на швидкість дифузійних процесів, ніж хімічних.
Залежність концентрації від часу для фіксованого перетину встановлюється за допомогою другого закону Фіка:
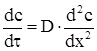 (ХІ.17.3)
(ХІ.17.3)
Дифузія має стаціонарний характер, коли концентрація змінюється в залежності від віддалі, а від часу не залежить
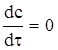 , (ХІ.17.4)
, (ХІ.17.4)
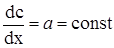 (ХІ.17.5)
(ХІ.17.5)
після інтегрування цього рівняння одержуємо
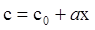 , (ХІ.17.6)
, (ХІ.17.6)
де с0 відповідає координаті х = 0.
З цього випливає, що при стаціонарній дифузії спостерігається лінійна зміна концентрації вздовж напрямку дифузії, а градієнт концентрації може бути записаний за допомогою кінцевих величин:
 , (ХІ.17.7)
, (ХІ.17.7)
де δ − кінцеве значення х. Підставляючи цей вираз у рівняння (ХІ.17.2), визначимо швидкість стаціонарної дифузії
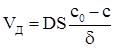 (ХІ.17.8)
(ХІ.17.8)
Іноді цю формулу записують іще так
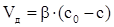 , (ХІ.17.9)
, (ХІ.17.9)
де β − коефіцієнт масопередачі.
Розглянемо гетерогенну хімічну реакцію, яка має перший порядок і протікає стаціонарно. Припустимо, що в цьому процесі можна виділити лише дві послідовні стадії: власне хімічну реакцію і процес дифузії, яким забезпечується необхідний транспорт реагентів до поверхні. (Для простоти вважаємо, що відведення продуктів реакції відбувається дуже швидко і його можна не враховувати).
Внаслідок стаціонарності даного процесу на поверхні розділу фаз не відбувається накопичення вихідних речовин або продуктів, тому швидкості обох стадій однакові. Із умови стаціонарності
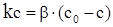 (ХІ.17.10)
(ХІ.17.10)
знаходимо
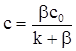 . (ХІ.17.11)
. (ХІ.17.11)
Підставимо це значення у вираз швидкості реакції і одержимо
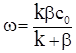 (ХІ.17.12)
(ХІ.17.12)
або
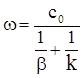 , (ХІ.17.13)
, (ХІ.17.13)
де 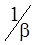 − дифузійний опір;
− дифузійний опір;  − хімічний опір.
− хімічний опір.
У випадку, коли k>>β, швидкість гетерогенної реакції рівна βс0 і визначається лише величиною β, що характеризує дифузію. Процес протікає в дифузійній області. У другому випадку β>>k w = kс0 сумарний процес визначається хімічною стадією і протікає в кінетичній області. Більш загальним є вплив на швидкість сумарного процесу обох стадій, що розглядаються, роль яких залежить від особливостей реакції. Так, при зниженні температури і перемішуванні вплив дифузії зменшується
Розглянемо розчинення твердих тіл і газів у рідинах. Експериментально встановлена така формула для швидкості розчинення твердого тіла в рідині
 , (ХІ.17.14)
, (ХІ.17.14)
де S − поверхня дотику твердого тіла з рідиною; с − концентрація речовини, що розчиняється в глибині рідини; сн − концентрація насиченого розчину; k − стала для даних умов величина.
У відповідності з цією формулою швидкість розчинення тим більша, чим більша поверхня дотику фаз і різниця між досягнутою в дану мить часу концентрацією і її максимально можливою величиною.
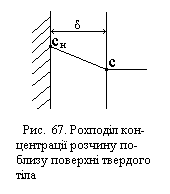 В. Нернст запропонував, що рідина біля поверхні твердого тіла утворює особливий, так званий дифузійний шар, за межами якого при доброму перемішуванні концентрація скрізь підтримується однаковою. Окрім того, він вважав, що швидкість самого розчинення значно більша швидкості дифузії, тому безпосередньо біля самої поверхні твердого тіла розчин близький до насичення.
В. Нернст запропонував, що рідина біля поверхні твердого тіла утворює особливий, так званий дифузійний шар, за межами якого при доброму перемішуванні концентрація скрізь підтримується однаковою. Окрім того, він вважав, що швидкість самого розчинення значно більша швидкості дифузії, тому безпосередньо біля самої поверхні твердого тіла розчин близький до насичення.
Зобразимо зміну концентрації розчиненої речовини в розчині графічно (рис. 67). Внаслідок того, що дифузія вважається найбільш повільною стадією, швидкість сумарного процесу можна прийняти за швидкість дифузії. Враховуючи умови стаціонарності і рівняння (ХІ.17.8), одержуємо
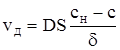 (ХІ.17.15)
(ХІ.17.15)
де δ − товщина дифузійного шару. Після об’єднання всіх сталих величин одержуємо рівняння (ХІ.17.14). Процес розчинення газу в рідинах у порівнянні з розчиненням твердих тіл відрізняється тим, що на межі розділу твердих фаз виникає два дифузійних шари. Один з них знаходиться біля поверхні розділу зі сторони газової фази, і другий − зі сторони рідини. Дифузійний шар, що розташований у газовій фазі, забезпечує підведення до поверхні рідини молекул одного з компонентів, що є в цій фазі. Перехід молекул компонента в глибину об’єму рідкої фази забезпечується другим дифузійним шаром, що розташований у рідині. В залежності від співвідношення швидкостей дифузії в цих шарах сумарний процес може лімітуватися в одному з шарів або визначатись обома шарами (рис.68).
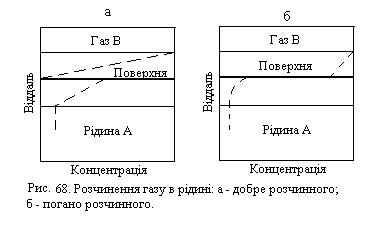 Для добре розчинного газу (рис. 68, а) градієнт концентрації в газовій фазі забезпечує граничну швидкість дифузії, яка може бути збільшена лише перемішуванням газу. При поглинанні малорозчинного газу, наприклад кисню у воді, граничного значення досягає градієнт концентрації в рідині і сумарний процес лімітується дифузією в рідкій фазі. Значить, в цьому випадку необхідно перемішувати рідину.
Для добре розчинного газу (рис. 68, а) градієнт концентрації в газовій фазі забезпечує граничну швидкість дифузії, яка може бути збільшена лише перемішуванням газу. При поглинанні малорозчинного газу, наприклад кисню у воді, граничного значення досягає градієнт концентрації в рідині і сумарний процес лімітується дифузією в рідкій фазі. Значить, в цьому випадку необхідно перемішувати рідину.
Теорія Нернста не в повній мірі відповідає дійсності. Обрахована на його основі товщина дифузійного шару виявляється настільки великою (порядку мільйона молекулярних шарів), що не можна його вважати таким, що не втягується в процес перемішування. В дійсності перенесення речовини між поверхнею розділу фаз і їх внутрішніми ділянками забезпечується не лише молекулярною дифузією, але і конвекцією, що пов’язана з рухом шарів речовини (конвективна дифузія). Тому розподіл концентрації біля поверхні розділу фаз не повинен підкорятися лінійному закону. Не дивлячись на те, що рівняння (ХІ.17.14) часто виконується, визначена з його допомогою величина 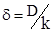 не відповідає дійсному приповерхневому шару і може бути названа лише ефективною товщиною дифузійного шару. Вона рівна товщині уявного дифузійного шару, який забезпечував би постачання речовин до поверхні розділу фаз лише шляхом молекулярної дифузії. Ефективна товщина дифузійного шару залежить від характеру руху фаз, що перемішуються (відсутність чи наявність завихрень, швидкості перемішування) і від властивостей речовин.
не відповідає дійсному приповерхневому шару і може бути названа лише ефективною товщиною дифузійного шару. Вона рівна товщині уявного дифузійного шару, який забезпечував би постачання речовин до поверхні розділу фаз лише шляхом молекулярної дифузії. Ефективна товщина дифузійного шару залежить від характеру руху фаз, що перемішуються (відсутність чи наявність завихрень, швидкості перемішування) і від властивостей речовин.
Дифузія біля поверхні реальних твердих тіл залежить від їх шершавості. Окрім зовнішньої дифузії, що відбувається в шарі рідини чи газу, які прилягають до твердого тіла, відбувається внутрішня дифузія в порах поверхні. Характер внутрішньої дифузії залежить від кількості, форми і розмірів пор. Для реакції, що протікає в дифузійній області, в залежності від температури, перемішування і будови поверхні роль лімітуючої стадії може виконувати як зовнішня, так і внутрішня дифузії.
У ряді випадків гетерогенні процеси як фізичної, так і хімічної природи пов’язані з утворенням нових фаз. При цьому фази, що раніше не існували, спочатку з’являються випадково, шляхом флуктуації у вигляді невеликих частинок (“зародків”), які лише в подальшому об’єднуються, утворюючи фазу достатньо великого об’єму. Стадія дрібнодисперсного стану пов’язана з утворенням великої поверхні нової фази і вимагає затрати енергії. Тому зародки нової фази виявляються менш стійкими як у порівнянні з вихідною фазою, так і з суцільною новою фазою достатньо великого об’єму. Якщо нова фаза термодинамічно більш стійка ніж стара, то існує деякий критичний розмір зародків, які можуть рости далі. Зародки меншого розміру, ніж критичний, мають тенденцію до зникнення.
В теорії кристалізації рідин розглядається імовірність ω утворення кристалічних зародків
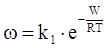 , (ХІ.17.16)
, (ХІ.17.16)
яка залежить від роботи W, що необхідна для їх утворення. Робота визначається коефіцієнтом поверхневого натягу σ і переохолодженням рідини ΔТ = Тпл − Т за рівнянням:
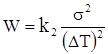 (ХІ.17.17)
(ХІ.17.17)
Окрім цього, використовується швидкість u подачі молекул рідкої фази до поверхні зародку
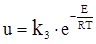 , (ХІ.17.18)
, (ХІ.17.18)
що залежить від енергії активації Е цього процесу.
Вважаючи, що швидкість v утворення кристалічних зародків пропорційна величинам ω і u:
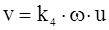 . (ХІ.17.19)
. (ХІ.17.19)
Поєднуючи рівняння (ХІ.17.16) і (ХІ.17.19) і вводячи замість k1, k2, k3, k4 і R нові коефіцієнти, одержуємо
 (ХІ.17.20)
(ХІ.17.20)
З рівнянням (ХІ.17.16) − (ХІ.17.20) випливає, що із зниженням температури плавлення зростає імовірність утворення зародків [ріст (ΔТ)2], але зменшується швидкість подачі молекул до поверхні зародків (ріст Е внаслідок збільшення в’язкості). Тому швидкість утворення кристалічних зародків із зниженням температури проходить через максимум.
З рівняння (ХІ.17.19) випливає, що швидкість утворення кристалічних зародків може бути мала при малих ω або u. Очевидно, утворення кристалів складної будови менш імовірне. Тому склоподібний стан більш властивий речовинам зі складною будовою кристалічної ґратки. Оскільки в’язкість рідини знижує швидкість подачі нових молекул до поверхні зародку, більш в’язкі рідини легше утворюють склоподібний стан. Утворенню склоподібного стану рідини сприяє швидке охолодження, при якому може бути досягнута велика в’язкість і одночасно перехід швидкості утворення зародків через максимум.
Теорія пояснює вплив на кристалізацію рідин добавок сторонніх речовин, які можуть слугувати центрами кристалізації, адсорбуватись на поверхні зародків, змінювати коефіцієнт поверхневого натягу та ін.
Поиск по сайту: