АвтоАвтоматизацияАрхитектураАстрономияАудитБиологияБухгалтерияВоенное делоГенетикаГеографияГеологияГосударствоДомДругоеЖурналистика и СМИИзобретательствоИностранные языкиИнформатикаИскусствоИсторияКомпьютерыКулинарияКультураЛексикологияЛитератураЛогикаМаркетингМатематикаМашиностроениеМедицинаМенеджментМеталлы и СваркаМеханикаМузыкаНаселениеОбразованиеОхрана безопасности жизниОхрана ТрудаПедагогикаПолитикаПравоПриборостроениеПрограммированиеПроизводствоПромышленностьПсихологияРадиоРегилияСвязьСоциологияСпортСтандартизацияСтроительствоТехнологииТорговляТуризмФизикаФизиологияФилософияФинансыХимияХозяйствоЦеннообразованиеЧерчениеЭкологияЭконометрикаЭкономикаЭлектроникаЮриспунденкция
ВВЕДЕНИЕ. Общие понятия и терминология процессов жидкостной экстракции
|
Читайте также: |
Общие понятия и терминология процессов жидкостной экстракции. Экстракция - процесс избирательного извлечения вещества из одной фазы в другую. Экстракцию газа жидкостью называют абсорбцией. Экстракция твердых веществ жидкими – выщелачивание. Экстракция растворенного вещества из одной жидкой фазы в другую носит название жидкостной экстракции или просто экстракцией.
Жидкостная экстракция – это процесс извлечения вещества из водного раствора в жидкую органическую фазу, не смешивающуюся с водой.
Перевод экстрагированного металла из органической фазы в водный раствор называется реэкстракцией.
Органическая фаза содержит органическое вещество (собственно экстрагент), образующее с извлекаемым металлом комплексные или солевые соединения, способные растворятся в органической фазе. Экстрагентами служат органические кислоты, спирты, эфиры, кетоны, амины и др.
Разбавитель – жидкое органическое вещество, не смешивающееся с водой, служащее растворителем экстрагента (керосин, ксилол, хлороформ, толуол и др.).
В качестве реэкстрагирующих растворов (реэкстрагентов) используют растворы кислот, солей и их оснований. В результате реэкстракции достигается и регенерация экстрагента. Органическая и водная фазы после экстракции называются соответственно экстрактом и рафинатом, а водный раствор после реэкстракции – реэкстрактом.
Количественной характеристикой экстракции является коэффициент распределения – К (отношение концентраций элемента в органической и водной фазах в состоянии равновесия):
 =
=  , где С1,С2,…Сi – концентрация элемента в различных его химических формах в органической и водной фазах.
, где С1,С2,…Сi – концентрация элемента в различных его химических формах в органической и водной фазах.
Когда в органической и водной фазах соединение экстрагируемого элемента имеет одинаковый состав, выражение упрощается: К = Кd = С(орг) / С(вод). Кd выражает закон распределения Нернста.
Селективность экстракции (избирательность) экстрагента по отношению к основному металлу (Ме1), которая для данной пары металлов (Ме1 и Ме2) определяется коэффициентом разделения: βме1/ме2 = К1/К2
Практически применяют экстракционные системы с β ≥ 2. Степень извлечения (ε) - процент извлечения металла в органическую фазу. При одинаковых объемах водной и органической фаз:  . При разных объемах водной Vвод и органической Vорг фаз:
. При разных объемах водной Vвод и органической Vорг фаз: 
Классификация экстракционных процессов. Нейтральные экстрагенты – органические вещества, молекулы которых образуют координационные связи с извлекаемым ионом.
Жидкие ионообменники - органические кислоты (основания) и их соли, способные при контакте с водным раствором к обмену неорганического катиона или аниона, входящего в состав экстрагента, на одноименный ион, находящийся в растворе.
Различают следующие типы механизмов экстракции: сольватный и гидратно-сольватный; анионообменный; катионообменный; простое физическое распределение; смешанный механизм экстракции.
Экстракция нейтральными экстрагентами. К нейтральным экстрагентам относят органические соединения, в составе которых имеются активные атомы, обладающие электроно – донорной способностью. Их можно подразделить на кислородсодержащие (активный атом >О::); азотсодержащие (активный атом >N:) и серосодержащие (активный атом = S::).
Равновесие экстракции. Экстракция трибутилфосфатом (ТБФ) с образованием сольвата описывается уравнением: Men+ + nNO3- + qТБФ ↔ Me(NO3)n qТБФ.
Константа равновесия реакции: Кс =  .
.
Так как [Me (NO3)n q ТБФ]орг / [Men+ ]вод = Кd, где Кd – коэффициент распределения, то
Кс = Кd / [ТБФ]qорг [NO3-]nводн.При [NO3-] = const, Kd = K' [ТБФ]qорг, где K' = Кс [NO3-].
Коэффициент распределения и полнота экстракции зависят от концентрации экстрагента.
Анионообменная экстракция – это экстракция соединений, находящихся в водном растворе в форме аниона. Анионообменные экстрагенты относятся к классу аминов, которые являются производными аммиака:
 Здесь R – углеводородный радикал, содержащий от 7 до 9 (иногда до 16) углеводородных атомов.
Здесь R – углеводородный радикал, содержащий от 7 до 9 (иногда до 16) углеводородных атомов.
Простейшим случаем экстракции является извлечение кислот, описываемое уравнением реакции нейтрализации: R3N+HXR ↔ [R3NH]X, где X - Cl -, SO42-, NO3- и др.
Образующиеся при обработке кислотой соли аминов могут обменивать анион кислоты на металлсодержащие анионы, например: 2[R3NH]Cl + PtCl62- ↔ [R3NH]2PtCl6 + 2Cl- .
Кроме анионного обмена, экстракция аминами иногда приводит к внедрению амина во внутреннюю координационную сферу экстрагируемого комплексного аниона с образованием прочных связей металл – азот.
В гидрометаллургии наиболее широко применяют третичные амины, в частности триалкиламин (ТАА) (С7Н15 ÷ С9Н17)3N. Амины экстрагируют металлсодержащие анионы только в кислых средах.
Равновесие анионообменной экстракции. При анионообменной экстракции коэффициент распределения элемента зависит от устойчивости комплексов, образующихся в водной фазе:
Кd = Кобм β АZ[S]n-z/F, где А – анион; n – заряд металлсодержащего аниона; Кобм – константа анионного обмена; β – константа устойчивости экстрагируемого комплекса; Z – степень окисления металла; F – функция закомплексованности (F = 1+ β1A+ β1A2+ β3A3+…); β1, β2, β3…– константы устойчивости комплексов МеА1, МеА2, МеА3 и т.д.
Полученный для хлоридных сред ряд экстрагируемости: Bi3+ > Sb3+ > Cd2+ > Zn2+ > Pb2+ > Fe3+>Cu2+>Co2+>Mn2+>Ni2+ совпадает с устойчивостью соответствующих хлорокомплексов.
Пример. В сернокислых растворах U (VI) может присутствовать в форме UO2SO4 и комплексных анионов UO2(SO4)22- и UO2(SO4)34-, соотношение между которыми определяется составом раствора. В зависимости от кислотности и концентрации ионов SO42- экстракция может протекать по типу присоединения или анионного обмена:
(R3NH)2SO4 + UO2SO4 ↔ (R3NH)2UO2(SO4)2 ,
(R3NH)2SO4 + UO2 (SO4)22- ↔ (R3NH)2UO2(SO4)2 + SO42-.
Катионообменная экстракция
К этому типу экстракции относится извлечение катионов металлов органическими кислотам, их солями и хелатообразующими реагентами.
Распространенными катионообменными экстрагентами являются кислоты жирного ряда типа RCOOH и их соли с числом углеродных атомов в цепочке радикала от семи до девяти, нафтеновые кислоты и их соли, разветвленные карбоновые кислоты.
Механизм экстракции состоит в обмене экстрагируемого металла на катион эктрагента, происходящем на границе раздела фаз.
Катионообменная экстракция описывается уравнением: Ме(вод)Z++zHR(орг)↔MeRz(орг)+zH+ (вод), где: Z – валентность; HR – органическая кислота; R – кислотный остаток органической кислоты.
Экстракция катиона кобальта органической солью («натриевым мылом») кислоты жирного ряда опишется: 2RCOONaорг + Cо2+вод ↔ (RCOO)2Cоорг + 2Na+ вод.
К катионообменным экстрагентам относятся также хелатообразующие реагенты – органические вещества, образующие с катионами металлов циклические комплексные соединения (внутрикомплексные соединения, или хелаты – от английского chelate compounds, т.е. клешневидные соединения). Молекулы хелатообразующих реагентов имеют в своем составе функциональные группы двух типов. Во-первых, это группы, из которых катион экстрагируемого металла вытесняет водород. Такими могут быть – СООН, – SО3Н, = РООН, однако значительно чаще хелатообразующие реагенты содержат гидроксильную (спиртовую) группу ≡ С–ОН или оксимную =NOH. Во-вторых, группы, образующие с металлом донорно-акцепторную связь. Эти группы содержат атомы кислорода (кетонная группа =С=О, гидроксильная –ОН) или азота (аминогруппы – NH2, = NH, ≡ N, оксимная группа =NOH или др.), обладающие повышенной донорной способностью.
Основные хелатообразующие реагенты – оксимы, оксиоксимы.
При взаимодействии хелатов с металлами, с одной стороны, как и в случае обычных катионообменных реагентов, происходит замещение протона функциональной группы реагента на экстрагируемый металл, с другой стороны – дополнительная координация металла донорными атомами молекулы экстрагента.
Важное достоинство хелатообразующих реагентов, особенно гидроксооксимов, – их высокая избирательность.
 Равновесие катионообменной экстракции. Экстракция катионов органической кислотой описывается уравнением: Мen+вод + n(HR)орг ↔ Me Rn орг + nH+вод. Константа равновесия реакции: Кс =
Равновесие катионообменной экстракции. Экстракция катионов органической кислотой описывается уравнением: Мen+вод + n(HR)орг ↔ Me Rn орг + nH+вод. Константа равновесия реакции: Кс =  .
.
В этом выражении  (коэффициент распределения).
(коэффициент распределения).
Поэтому Кс = Кd·[H+]nвод / [HR]nорг и Кd = Кс· [HR]nорг / [H+]nвод. Логарифмируя, получаем: lg Kd= lgKc + nlg[HR]орг + npH, поскольку рН = -lg[Н+]. В процессах экстракции, как правило, [HR]орг>>[Ме n+]вод, тогда [HR]орг ≈ const, тогда lg Kd = lg K' + npH,
где K' = Кс [HR]nорг. Таким образом, lg Kd является линейной функцией рН и не зависит от концентрации катионов [Ме n+]. Наклон прямой определяется зарядом катиона (рисунок 2). Для увеличения полноты экстракции необходимо связать выделяющиеся катионы Н+ в воду.
Умножив и разделив выражения для Кс на [ОН-]n, получим:
Кс =  .
.  , или Кс =
, или Кс =  , где: КW = аН+ · аОН– – константа воды; LMe(ОН)n – произведение растворимости гидроксида извлекаемого металла.
, где: КW = аН+ · аОН– – константа воды; LMe(ОН)n – произведение растворимости гидроксида извлекаемого металла.
Учитывая, что [НR]орг ≈ const, [МеRn]орг ≈ const, получим K' =  .
.
Подставив это выражение в уравнение для Kd, получим: lgKd = lgKc – lgLMe (ОН)n + n·рН. То есть, при прочих равных условиях, чем меньше L гидроксида, тем больше коэффициент распределения. По величине LMe(ОН)n ряд экстрагируемости металлов можно представить в виде:

Каждый стоящий в ряду металл, находящийся в водном растворе в виде соли минеральной кислоты, вытесняет из органической фазы все стоящие правее его металлы, находящиеся в виде соли органической кислоты. Например, ZnSO4вод + CuR2орг ↔ ZnR2орг + CuSO4вод .
Наиболее полная экстракция наблюдается в области рН гидратообразования. Экстракционный ряд (последовательность экстракции) совпадает с рядом металлов в порядке возрастания их рН гидролиза.
Экстрагируемость снижается при переходе от Ме3+ к Ме2+ и Ме+. В ряду катионов с одинаковым зарядом существенное значение имеют различия в энергиях гидратации ионов.
На полноту катионообменной экстракции также влияют:
– тип аниона минеральной кислоты (при прочих равных условиях Kα (NO3-) > Kα (Cl-) > Kα (SO42-));
– солевой фон раствора (чем выше солевой фон раствора, т.е. чем больше плотность раствора, тем меньше Kd).
При экстракции мылами происходит обмен катионами металлов между органической и водной фазами: Me1n+ + Me2Rn = Me1Rn + Me2n+. Константа равновесия реакции:  .
.
Если Кd(ме1)>Кd(ме2), реакция идет в прямом направлении, если Кd(ме2) > Кd(ме1) – в обратном.
Физическое распределение. Данный тип экстракции наблюдается, когда извлекаемое соединение координационно насыщенно, а поэтому не гидратированно и не диссоциирует в водной фазе, оно извлекается в органическую фазу без образования комплексов.
Это относительно редкий для экстракции неорганических соединений случай, когда экстракция не сопровождается химическим взаимодействием. Экстракция происходит за счет действия ван-дер-ваальсовых сил или слабого донорно-акцепторного взаимодействия. При этом диссоциация извлекаемого вещества в водной фазе не велика или подавлена введением специальных реагентов.
Экстракция смесями экстрагентов. Органическая фаза экстракционных систем, как правило, является раствором экстрагента в разбавителе. В качестве разбавителя используют углеводороды (предельные, ароматические, циклические). В качестве добавок – модификаторов используют органические спирты, кетоны или ТБФ. Из перечисленных веществ только углеводороды сравнительно слабо взаимодействуют с компонентами органической фазы. Остальные разбавители обладают сольватирующей способностью, а некоторые из них имеют существенную экстракционную способность. Поэтому при использовании смесей органических веществ наблюдаются отклонения от аддитивности экстракции. Это явление получило название синергизма или синергетного эффекта. Синергизм наблюдается во многих экстракционных системах и обусловлен химическими взаимодействиями в органической фазе.
Эффект смешения экстрагентов (ЭС) равен: ЭС = Dэксп/Dрасч, где: Dэксп и Dрасч – найденные экспериментально и вычисленные (из предположения аддитивности) коэффициенты распределения.
Если ЭС = 1, синергентный (или антисинергентный) эффект отсутствует; при ЭС> 1 имеет место синергентный эффект, при ЭС< 1 – антисинергентный эффект.
Синергентные эффекты наблюдаются в ряде систем с солеобразующим (органические кислоты, амины) и нейтральным (ТБФ, кетоны) экстрагентами, с двумя нейтральными экстрагентами, со смесью катионообменного и анионообменного экстрагентов, в системах хелатный – нейтральный экстрагенты.
Пример. В системах катионообменный экстрагент (кислота НR) – нейтральный экстрагент (S) синергизм может быть обусловлен замещением сольватно связанных молекул НR молекулами S: MeRn · mHR + mS ↔ MeRn · mS + mHR.
Выделение свободного экстрагента HR, который обычно экстрагирует лучше, чем нейтральный, приводит к резкому увеличению коэффициента распределения (иногда в 1000 – 10000 раз).
Кроме того, синергизм в системе органическая кислота – нейтральный экстрагент объясняется возможностью дополнительного присоединения нейтрального экстрагента по реакции: Men+вод + n(HR)орг + mS ↔ Me(R·HR)nSm орг+nH+вод. В этом случае вследствие повышения координационного числа и образования более крупной молекулы возрастает коэффициент распределения металла.
При переходе воды в органическую фазу синергизм может быть вызван вытеснением сольватно связанной воды молекулами нейтрального экстрагента:
MeRn·mH2O+mS↔MeRn·mS + mH2O.
Аналогичная реакция может иметь место при экстракции смесью анионообменного и нейтрального экстрагентов. Кроме того, синергетный эффект может быть обусловлен присоединением молекул нейтрального экстрагента к анионному комплексу металла. Так, при экстракции молибдена из солянокислых растворов смесью ТОА и ТБФ протекает реакция:
MoO2Cl2 + [R3NH]Cl+ТБФ ↔ [ R3NH]+[ MoO2Cl3 · ТБФ]-.
При экстракции смесями анионообменного и катионообменного экстрагентов синергетный эффект наблюдается в результате замещения в комплексе неорганического аниона на органический, что приводит к образованию лучше экстрагируемого соединения:
[AmH+][MeYX]– + yHR ↔ [AmH] +[MeYx-yRy]– + yHY, где: HR – органическая кислота; HY – минеральная кислота; [АmH+]- – органический катион (алкиламмоний).
Кинетика процессов экстракции. Скорость экстракции зависит от скорости массопередачи и от скорости химических реакций внутри обеих фаз на межфазной границе.
В условиях интенсивного перемешивания в экстракционных аппаратах скорость массопередачи достаточно высока. В некоторых случаях низкая скорость экстракции объясняется не медленным образованием экстрагируемого соединения, а другими химическими реакциями, например, гидролизом или комплексообразованием в водной фазе, предшествующими экстракции.
Общее уравнение скорости экстракции, учитывающее диффузионные и химические сопротивления, описывается выражением: j = (CR – CR*) / (1/βR + 1/k βE +1/k1), где: CR – средняя концентрация распределяемого компонента в рафинате; CR* – концентрация в объеме рафината, равновесная со средней концентрацией компонента в экстракте; βR и βE – коэффициенты массопередачи в фазах; k – коэффициент распределения элемента; k1 – константа скорости прямой реакции (экстракции).
Примеры экстракция в гидрометаллургии. Получение меди, получение редких металлов, получение урана.
Ионный обмен – реакция обмена ионами ионита и раствора, обмен ионов между двумя электролитами:  , или
, или  , где: R и R1 – матрицы катионита и анионита; Н+ и ОН- - противоины. (Чертой отмечена твердая фаза).
, где: R и R1 – матрицы катионита и анионита; Н+ и ОН- - противоины. (Чертой отмечена твердая фаза).
Иониты часто используют не в виде кислот или оснований, а «заряжают» в солевую форму, например, катиониты -  или
или 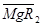 , аниониты -
, аниониты - 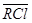 ,
, 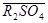 и т.п. В этом случае противоионами, обменивающимися с коионами электролита являются Na+, Mg2+, Cl- и SO42-.
и т.п. В этом случае противоионами, обменивающимися с коионами электролита являются Na+, Mg2+, Cl- и SO42-.
Ионообменные процессы применяют для решения ряда задач: 1) селективного извлечение металлов из бедного раствора; 2) концентрирования раствора извлекаемого металла; 3) разделение близких по свойствам элементов: Zr и Hf, Ni и Co и др.; 4) получение высокочистой и умягчённой воды; 5) очистка от примесей различных производственных растворов, обезвреживание сточных вод.
Типы ионообменных соединений. Ионообменники могут быть разделены на группы по составу – на неорганические и органические; по методу приготовления – на природные и синтетические.
Неорганические иониты. Природные минеральные сорбенты, обладают, как правило, катионообменными свойствами. Это полевые шпаты, глинистые минералы, производные различных смол, цеолиты (алюмосиликаты) и др. Катионообменные свойства этих веществ обусловлены, как правило, алюмосиликатами типа Na2O . Al2O3 . nSiO2 . mH2O (где n и m – переменные).
Цеолиты, например, построены из чередующихся кремнекислородных (SiO44-) и алюмокислородных (AlO45-) тетраэдров. Они соединяются в вершинах общими кислородными атомами. В силикатной решётке часть ионов Si4+ замещена Al3+. Вследствие этого решётка цеолита несёт отрицательный электрический заряд. Он компенсируется положительным зарядом щелочного или щелочноземельного металлов. Их ионы не занимают фиксированных положений и свободно передвигаются в структуре решётки. Благодаря присутствию в решётке подвижных положительных ионов цеолиты обладают способностью обменивать катионы.
Природные минеральные сорбенты обычно применяют в процессах водоподготовки. Их достоинства – высокая термическая и радиационная стойкость и дешевизна. Недостатки – малая поглотительная способность и невысокая механическая прочность.
К синтетическим неорганическим сорбентам относятся искусственно полученные соединения: силикагели, пермутиты, труднорастворимые оксиды и гидроксиды ряда металлов (алюминия, хрома, олова, циркония, титана и др.).
Силикагель – диоксид кремния с различным количеством молекул воды: SiO2·nH2O. Катионообменная способность силикагелей проявляется в щелочной среде путем обмена водорода гидроксильных групп на катионы металлов.
Пермутиты – катионообменные сорбенты – искусственные алюмосиликаты. Получают их сплавлением соединений, содержащих алюминий и кремний.
В отличие от природных минеральных сорбентов обладают большей ёмкостью.
Органические иониты. Природные - почвы, торф, некоторые угли, продукты сульфирования и фосфорирования угля, торфа, лигнина и целлюлозы различного происхождения (дерево, бумага, хлопок, ткань, фруктовая косточка). Природные вещества обычно применяют после предварительной химической обработки.
В металлургии широко распространены синтетические ионообменные смолы – высокомолекулярные пористые полимеры. Они состоят из эластичной трёхмерной сетки углеводородных цепей, в которых закреплены фиксированные ионы: у катионитов – SO3 -, – COO -, – PO32-, – AsO32 - и др., у анионитов – NH3 +, = NH2 +, ≡ NH +, ≡ N +.
Фиксированный ион связан с противоионом и образует с ним ионогенную группу, которую часто называют активной или функциональной группой. Активными группами являются: – SO3Н, – SO3Na, – COOH, – PO3H2, – AsO3Na2, – NH3Cl, ≡ NOH и др.
От степени диссоциации активных групп зависит способность смолы к ионному обмену.
В связи со способностью смол образовывать ионы в растворах электролитов (процесс диссоциации) их в зависимости от степени диссоциации делят на слабые и сильные. Сильнокислотными считают катиониты, содержащие группы – PO3H2, – SO3H, (сульфокатионы), а слабокислотными – фенольные группы ОН - и группы СООН (карбоксильные катиониты).
Сильноосновные аниониты содержат группы, например NR3OH, хорошо диссоциирующих четвертичных аммониевых или пиридиновых оснований. Подобные смолы способны к обмену анионов не только в кислых, но и щелочных средах. Слабоосновные аниониты содержат первичные, вторичные и третичные аминогруппы, которые являются слабыми основаниями и диссоциируют лишь при pH ниже 7. Смолы могут быть полифункциональными, т.е. содержать однлвременно различные активные группы. При этом катиониты содержат функциональные группы различной кислотности, аниониты – группы различной основности, а амфолиты содержат как кислотные, так и основные функциональные группы.
Ионообменные смолы проявляют селективность к ионам. Большей избирательности ионообменных смол можно добиться путём введения в их структуру дополнительных групп, образующих с ионами координационную связь. Так, например, в результате введения в структуру смолы глиоксима получен катионит, селективно сорбирующий никель; введение в смолу остатка 8 – оксихинолина сообщает смоле избирательность в отношении сорбции ионов кобальта.
Ионогенные группы гидрофильны. При контакте с водой эти группы стремятся раствориться. Нерастворимость смол в водном растворе достигается введением поперечных связей для связывания углеводородных цепей.
Содержание поперечных связей, или так называемая степень сшивки, определяет физические свойства смолы. При контакте с водой ионит в большей или меньшей мере набухает, но остаётся нерастворимым в воде и в других растворителях.
Для синтеза ионообменных смол используют процессы полимеризации и/или поликонденсации мономерных молекул органических соединений. Это взаимодействие приводит к образованию пространственной решётки углеводородных цепей молекул.
Полимеризационные катиониты представляют собой сетчатые полимеры стирола с поперечными связями, в которые кислотной обработкой вводятся кислотные группы. Мономеры стирола сополимеризуются с различным количеством дивинилбензола, служащим мостикообразователем.
Широко распространены смолы на основе стирола и дивинилбензола. Гранулы сополимера обрабатывают концентрированной серной кислотой (сульфируют), при этом образуются сульфокислоты – катиониты с ионогенной сульфогруппой – SO3H:
Электроноионообменники или редокссмолы (ЭИ) – это высокомолекулярные поперечносвязанные не растворимые в воде вещества, в которых имеются группы, способные к обратимому окислению или восстановлению, т.е. способные к отдаче или получению электронов.
Мембраны – это синтетические органические иониты, приготовленные в форме листов. Когда ионообменная мембрана соприкасается с раствором электролита, то противоионы без труда проходят через мембрану от одного раствора к другому. Соионы на другую сторону не проникают.
Основные характеристики ионообменных смол
Набухаемость материалов. Так как ионы присутствуют в растворе в форме гидратированных, то при поглощении сорбентом иона вместе с ним в структуру ионита проникает гидратная вода, что приводит к набуханию. Набухание сопровождается растяжением пространственной сетки смолы и увеличением её объёма. Набухаемость зависит от числа ионогенных групп и поперечных связок. С увеличением числа поперечных связок набухаемость уменьшается (возрастает жёсткость каркаса).
Набухание характеризуется коэффициентом набухания:  , где VH – удельный объем набухшей смолы; Vc – удельный объем смолы в воздушно-сухом состоянии.
, где VH – удельный объем набухшей смолы; Vc – удельный объем смолы в воздушно-сухом состоянии.
Емкость ионитов. Обменная емкость ионита определяется числом активных групп, в миллиграмм – эквивалентах (мг·экв), приходящихся на один грамм или миллиграмм сорбента.. Ёмкость ионита зависит от валентности сорбируемого иона – чем больше валентность, тем больше активных центров будет занята одним ионом, и тем меньше обменная ёмкость
Различают полную обменную ёмкость (ПОЕ), статическую (равновесную) обменную ёмкость (СОЕ) и динамическую обменную емкость (ДОЕ). Для практических целей используют понятие о рабочей обменной ёмкости, соответствующей конкретным рабочим условиям. СОЕ в конкретных условиях зависит от суммы факторов, влияющих на состояние равновесия.
Полная обменная ёмкость (ПОЕ) - Спол - количество сорбированных ионов при заполнения всех активных центров ионита.
Динамическая ёмкость (ДОЕ) - Сдин - количество ионов, сорбированных единицей массы сорбента в динамическом режиме (фильтрацией) до проскока сорбируемого иона через слой смолы.
Статическая ёмкость (СОЕ) - Сстат - количество ионов, сорбированных единицей массы смолы, находящейся в статическом контакте с раствором данного электролита в принятых условиях.
Рабочая ёмкость (Сраб) выражает количество ионов, сорбированных единицей массы (или объёма) смолы в конкретных технологических условиях.
Избирательность смол. Избирательность ионитов зависит от степени сродства иона к иониту, а также от степени сшивки синтетических органических ионитов (размер пор ионитов) – чем больше степень сшивки, тем сложнее крупным ионам проникать в смолу. Многовалентные ионы сорбируются, как правило, более избирательно, чем ионы с меньшей валентностью.
Действие комплексообразователей. Если ионы образуют комплексы с компонентами раствора, ёмкость сорбента изменится.
Прочность ионитов. Механические нагрузки связаны с трением зёрен ионита между собой и о стенки аппарата, ударами о частицы руды. На прочность ионитов влияет степень сшивки: при малой степени сшивки ионит при набухании может превратиться в гелеобразную массу, при жёсткой сшивке в процессе сорбции за счёт возрастающих внутренних напряжений ионит может разрушиться.
На ионит могут оказывать воздействие химические вещества, содержащиеся в растворах.
На прочность ионитов влияет температура. Их не рекомендуют применять при температуре выше 60 ºС.
Жёсткое облучение в ряде случаев существенно влияет на свойства ионитов.
Равновесие ионного обмен. Реакцию извлечения при ионном обмене можно записать так:  ,где А и В – обменивающиеся ионы; ZA и ZB – их заряды; ν – стехиометрические коэффициенты; R – одновалентный противоион извлекающегося вещества.
,где А и В – обменивающиеся ионы; ZA и ZB – их заряды; ν – стехиометрические коэффициенты; R – одновалентный противоион извлекающегося вещества.
По условию электронейтральности электролита  νA; откуда:
νA; откуда: 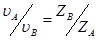 . Тогда можем записать:
. Тогда можем записать:  ,или в ионной форме:
,или в ионной форме:  . Константы равновесия реакций будут иметь вид:
. Константы равновесия реакций будут иметь вид: 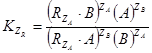 , и
, и  , где
, где  ,
,  , aA и aB – активности ионов в смоле и в растворе.
, aA и aB – активности ионов в смоле и в растворе.
При отсутствии данных об активностях ионов, используют соответствующие концентрации и применяют концентрационную константу равновесия: 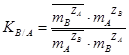 , где где
, где где  ,
, 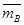 , aA и aB – моляльные концентрации ионов в смоле и в растворе. Разделив на (ZA . ZB), получим:
, aA и aB – моляльные концентрации ионов в смоле и в растворе. Разделив на (ZA . ZB), получим:  , откуда
, откуда 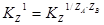 .
.
Для оценки процесса сорбции используют коэффициент распределения D икоэффициент разделения  - коэффициент селективности:
- коэффициент селективности:
 ;
; 
 Изотермы ионного обмена. Изотерма выражает зависимость между эквивалентной долей противоиона А в ионите (
Изотермы ионного обмена. Изотерма выражает зависимость между эквивалентной долей противоиона А в ионите ( ) и эквивалентной долей его в растворе (
) и эквивалентной долей его в растворе ( ) в состоянии равновесия реакции:
) в состоянии равновесия реакции:  -
-  и
и  где: m – концентрация в моляльностях, Z1 и Z2 – заряды обменивающихся ионов.
где: m – концентрация в моляльностях, Z1 и Z2 – заряды обменивающихся ионов.
Следовательно, произведения  ,
,  ,
,  и
и  равны числу эквивалентов в единице массы растворителя или ионита.
равны числу эквивалентов в единице массы растворителя или ионита.
При отсутствии избирательности соотношение концентраций ионов А и В в ионите и растворе одинаково. В этом случае изотерма изображается прямой с наклоном 45° (пунктирная линия на рисунке 1). При избирательном поглощении ионов А изотерма проходит выше диагонали квадрата -1, при избирательном поглощении ионов В - ниже диагонали -2. Селективность ионита обусловливает нелинейность изотермы ионного обмена.
Коэффициент разделения  равен отношению площадей прямоугольников под и над изотермой, соприкасающихся с ней в точке, отвечающей заданному составу (рис. 1),
равен отношению площадей прямоугольников под и над изотермой, соприкасающихся с ней в точке, отвечающей заданному составу (рис. 1),  ;
; 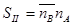 .
. 
Кинетика ионного обмена.
Общие положения. Ионообменный процесс включает пять стадий: 1) диффузию ионов через раствор электролита к поверхности зерна ионита; 2) диффузию ионов внутри зерна; 3) протекание самой реакции ионообмена; 4) диффузию вытесненного противоиона изнутри к поверхности ионита; 5) диффузию вытесненного противоиона от поверхности ионита в объём раствора.
Стадии 1 и 5, 2 и 4 по сути одинаковы, только противоположны по направлению. Общая скорость полного обмена определяется скоростью процесса, протекающего медленнее (химической реакцией или диффузией). Скорость ионообменных реакций на смолах регулируется диффузией, являющейся самой медленно протекающей стадией.
Различают диффузию в зоне ионита (гелевая диффузия) и диффузию через плёнку жидкости, примыкающую к поверхности (плёночная диффузия). В некоторых случаях обе стадии могут контролировать процесс. Большей частью в концентрированных растворах (выше 0,1 М) скорость обмена контролируется диффузией в зерне, при низких концентрациях – внешней диффузией.
Строение ионита также влияет на величину коэффициента диффузии. Малонабухающие иониты с высоким содержанием мостикообразующего материала хуже пропускают противоионы внутрь зерна. На коэффициент диффузии влияет также и заряд ионита. Коэффициент диффузии для крупных ионов будет меньше, чем для ионов небольших размеров.
На коэффициент внутренней диффузии влияет концентрация раствора электролита. В разбавленных растворах благодаря высокой ёмкости ионита скорость ионного обмена определяется диффузией в растворе даже в случае интенсивного перемешивания.
При увеличении температуры коэффициент диффузии растёт и соответственно повышается скорость ионообменной реакции. Однако, при температуре более 50 – 60оС большинство смол разрушается.
Закономерности плёночной кинетики. При плёночной кинетике скорость процесса определяется в соответствии с первым законом Фика. Для зерна сферической формы можно записать:  , где: q – количество сорбированного противоиона; r0 – радиус частицы смолы;
, где: q – количество сорбированного противоиона; r0 – радиус частицы смолы;  – толщина плёнки; D – коэффициент диффузии; С – концентрация противоиона в растворе; Спов – концентрация противоиона на поверхности.
– толщина плёнки; D – коэффициент диффузии; С – концентрация противоиона в растворе; Спов – концентрация противоиона на поверхности.
Общее количество сорбированного зерном противоиона равно: 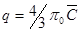 , где
, где  – концентрация противоиона в смоле.
– концентрация противоиона в смоле.
В состоянии равновесия  , где α – коэффициент распределения.
, где α – коэффициент распределения.
Подставим Спов в уравнение Фика:  .
.
Количество сорбированного противоиона при равновесии определяется уравнением:  .
.
 α и, т.к. при насыщении
α и, т.к. при насыщении  , то
, то  .
.
Преобразуя уравнение Фика, получим  , где
, где  .
.
Интегрируя полученное уравнение при граничных условиях q = 0 при τ = 0, получим: 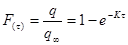 , где F(τ) –доля сорбированного иона от максимально возможного (равновесного) значения.
, где F(τ) –доля сорбированного иона от максимально возможного (равновесного) значения.
После логарифмирования уравнение приводится к удобной форме:  .
.
Закономерности гелевой кинетики. Для определения коэффициента диффузии используют 2-е уравнение Фика.  , где: F(τ) = q/q∞ – отношение продиффундировавшего количества вещества за время τ (q) к равновесному количеству сорбированного вещества (q∞);
, где: F(τ) = q/q∞ – отношение продиффундировавшего количества вещества за время τ (q) к равновесному количеству сорбированного вещества (q∞);  – коэффициент диффузии, см2/сек; n – количество слоёв шарообразной частицы; τ – время, сек; r – радиус, см.
– коэффициент диффузии, см2/сек; n – количество слоёв шарообразной частицы; τ – время, сек; r – радиус, см.
Скорости диффузии ионов внутри смолы много меньше скорости диффузии их в растворе.
 Построение изотерм сорбции. Изотермы сорбции необходимы для определения ёмкости сорбента и числа ступеней сорбции для извлечения элемента из раствора. Изотермы сорбции строят в виде зависимости между равновесным содержанием элемента в сорбенте Е (мг/г, мг/экв/г) и равновесной концентрацией его в растворе (мг/мл, г/дм3, мг/экв/дм3) (рисунок 2).
Построение изотерм сорбции. Изотермы сорбции необходимы для определения ёмкости сорбента и числа ступеней сорбции для извлечения элемента из раствора. Изотермы сорбции строят в виде зависимости между равновесным содержанием элемента в сорбенте Е (мг/г, мг/экв/г) и равновесной концентрацией его в растворе (мг/мл, г/дм3, мг/экв/дм3) (рисунок 2).
Наибольшей селективностью к поглощаемому иону при достаточно высокой ёмкости обладает ионит 1. Резкий подъём изотермы сорбции иона означает возможность достаточно полного его извлечения из растворов с низким содержанием. Ионит 2 менее селективен и достигает высокой ёмкости лишь при значительных концентрациях иона в растворе. Ионит 3 обладает несколько меньшей ёмкостью в растворах с высокой равновесной концентрацией, чем предыдущие иониты. Изотерма 4 скорее характеризует процесс физической сорбции.
Изотермы позволяют графическим методом рассчитать число ступеней сорбции (рисунок 3.).
 Для расчёта задаются степенью извлечения иона, т.е. конечной концентрацией его в растворе - Ск. Исходная концентрация иона в растворе - Сн. Степень насыщения ионита Е обычно составляет 90 – 98 % от максимальной его ёмкости Е. При элюировании не достигают полного извлечения поглощённого элемента. Поэтому после элюирования ионит сохраняет остаточную ёмкость – Ен. Она является начальной ёмкостью перед новой загрузкой. Пересечение перпендикуляра из точек Ек и Сн даёт точку А, а из точек Ен и Ск – точку N. Прямая, соединяющая точки А и N является рабочей линией.
Для расчёта задаются степенью извлечения иона, т.е. конечной концентрацией его в растворе - Ск. Исходная концентрация иона в растворе - Сн. Степень насыщения ионита Е обычно составляет 90 – 98 % от максимальной его ёмкости Е. При элюировании не достигают полного извлечения поглощённого элемента. Поэтому после элюирования ионит сохраняет остаточную ёмкость – Ен. Она является начальной ёмкостью перед новой загрузкой. Пересечение перпендикуляра из точек Ек и Сн даёт точку А, а из точек Ен и Ск – точку N. Прямая, соединяющая точки А и N является рабочей линией.
Точки А и N должны находиться внутри выпуклой изотермы. Далее строят ломаную линию АВDKLMN. Она состоит из отрезков прямых, параллельных абсциссе (до пересечения с изотермой) и ординате (до пересечения с рабочей линией).
Число вертикальных (горизонтальных) отрезков соответствует необходимому числу ступеней сорбции при работе в статических условиях. В данном случае три ступени сорбции.
Ионообменная хроматография. Для разделения близких по свойствам металлов в ряде случаев используют ионообменную хроматографию. Метод основан на различном сродстве к ионообменной смоле ионов, содержащихся в протекающем через слой ионита растворе.
При фронтальном способе ионообменной хроматографии исходный раствор непрерывно поступает в верхнюю часть ионообменной колонки. Так как скорости перемещения ионов каждого вида различны, то в вытекающем снизу колонки растворе будет больше тех ионов, которые имеют меньшее сродство к смоле.
При вытеснительной или элюентной хроматографии разделяемые ионы сначала совместно сорбируются смолой в верхней части колонки. Затем вымываются (элюируются) раствором, содержащим элюирующий реагент (десорбент). При вымывании по длине колонки образуются зоны, которые содержат ионы определённого вида и перемещаются вдоль колонки с различной скоростью.
При вытеснительной хромаграфии элюирующие ионы имеют сродство к смоле больше, чем разделяемые ионы и ион, которым первоначально заряжена смола.
При вытеснительной хроматографии (см. схемы) в верхней части смолы, заряженной ионом А, происходит сорбция ионов В и С.


Затем при воздействии элюирующего раствора D произойдет вытеснение ионов В и С. Это вызовет движение полосы сорбции В и С. Если Dc > DВ, то внутри полосы будет происходить перераспределение. В передней части полосы концентрируются ионы В, а в хвостовой - С. В средней части находится зона, содержащая смесь В+С. Вытекающий раствор содержит поочередно АУ, затем ВУ, СУ и ДУ (У – противоион соединения в растворе). Полоса сорбции не меняет свою ширину. В пограничных слоях будут содержаться смеси ионов А+В, В+С, С+Д, т.к. слои вплотную прижаты друг к другу.
При элюентной хроматографии вымывание ионов проводят раствором, содержащим ион, имеющий меньшее сродство к смоле, чем разделяемые ионы.
Электродиализ. В последнее время значительное развитие получила сорбция с использованием ионитовых мембран – электродиализ. Мембраны – это тонкие гибкие листы. Изготавливают их из ионообменных смол. Они подразделяются на гомогенные и гетерогенные. Гомогенные состоят только из ионообменных смол. Гетерогенные – мембраны, в состав которых входят смолы и связующие вещества, армирующая ткань для создания прочности и эластичности мембран. Мембраны электропроводны. Очень важным электрохимическим свойством мембран является их селективность - они проницаемы только для ионов, имеющих заряд того же знака, что и у подвижных ионов смолы.
Селективности мембран зависит от величины мембранного потенциала. Потенциал возникает между растворами, которые разделены мембраной. По Нернсту  , где UK и UA – подвижности катиона и аниона, см2/с; z – заряд иона; F – число Фарадея; а1 и а2 – активности по обе стороны мембраны.
, где UK и UA – подвижности катиона и аниона, см2/с; z – заряд иона; F – число Фарадея; а1 и а2 – активности по обе стороны мембраны.
Для катионитовой мембраны идеальной селективности UA ≈ 0; для идеальной анионитовой UK ≈ 0. Поэтому для идеальной мембраны первый множитель уравнения равен единице.
Лекция 15. Кинетика и механизм гидрометаллургических процессов. Электролитическое осаждение и рафинирование металлов
Общее уравнение массообмена при выщелачивании. Выщелачивание твердого тела - гетерогенный процесс. Процесс состоит из трех основных стадий: диффузия исходных веществ; химические превращения; диффузия продуктов реакции. Если в ходе процесса образуется новая твердая фаза, то диффузионные стадии включают кроме внешней – внутреннюю диффузию.
Схематическая модель выщелачивания приведена на рисунке 1.
Уравнение потока процесса выщелачивания имеет вид –
j =  ,
,
где: j – поток вещества в установившемся режиме; D1 – коэффициент внешней диффузии; D2 – коэффициент внутренней диффузии; К – константа скорости кристаллохимических превращений.
 - коэффициент массопередачи, а
- коэффициент массопередачи, а  - диффузионное сопротивление. Величина 1/К - химическое сопротивление. Общее сопротивление складывается из диффузионных и химических сопротивлений:
- диффузионное сопротивление. Величина 1/К - химическое сопротивление. Общее сопротивление складывается из диффузионных и химических сопротивлений:  .
.
 Закономерности и признаки внешней диффузии. Различают молекулярную (концентрационную) и конвективную диффузии.
Закономерности и признаки внешней диффузии. Различают молекулярную (концентрационную) и конвективную диффузии.
Молекулярная диффузия обусловлена наличием разности концентрации вещества у поверхности твердой фазы и в глубине раствора и определяется 1 законом Фика: количество вещества, прошедшее в результате диффузии через плоскую поверхность, пропорционально величине поверхности, продолжительности диффузии и градиенту концентрации в направлении, перпендикулярном поверхности:
 ,
,
где: D – коэффициент диффузии; S – площадь; dC/dX – градиент концентрации в направлении оси Х, перпендикулярной поверхности.
Удельное количество вещества, подводимого с потоком жидкости (конвекцией), определяется выражением: j = V (CO – C a).
Если скорость лимитируется внешней диффузией, то уравнение потока имеет вид:  .
.
Основные признаи протекания процесса во внешнедиффузионном режиме:
1) Скорость процесса находится в линейной зависимости от концентрации реагента, поскольку  (коэффициент внешней массопередачи).
(коэффициент внешней массопередачи).
2) Скорость процесса зависит от скорости движения жидкости относительно поверхности твердой фазы, так как  . Чем больше скорость движения жидкости, тем меньше величина dд и, значит, скорость диффузии больше. Этот признак является определяющим: влияние скорости перемешивания на скорость выщелачивания всегда указывает на лимитирование скорости процесса внешней диффузией.
. Чем больше скорость движения жидкости, тем меньше величина dд и, значит, скорость диффузии больше. Этот признак является определяющим: влияние скорости перемешивания на скорость выщелачивания всегда указывает на лимитирование скорости процесса внешней диффузией.
3) Скорость процесса не зависит от времени, если СО = const, так как при V = const,  и внешнее диффузионное сопротивление постоянно во времени.
и внешнее диффузионное сопротивление постоянно во времени.
4) Скорость процесса мало зависит от температуры, так как коэффициент диффузии и вязкость раствора незначительно изменяется с температурой.
Кажущаяся энергия активации процесса мала и составляет порядка 10 – 20 кДж/моль.
Закономерности и признаки внутридиффузионного режима выщелачивания. Скорость внутренней диффузии определяется физико-химическими свойствами нерастворимой твердой фазы, покрывающей растворяющееся вещество и, в первую очередь, плотностью этой оболочки: чем выше плотность оболочки, т.е. меньше ее пористость, тем больше затруднена диффузия через оболочку и тем меньше скорость внутренней массопередачи.
Ориентировочное суждение о плотности оболочки продукта может быть сделано на основании величины критерия Пиллинга-Бедвордса, который определяется как отношение молярных объемов твердого продукта реакции и исходного твердого вещества.
Если КП-Б < 1, то молярный объем продукта реакции меньше молярного объема исходного вещества, оболочка будет не сплошная, рыхлая и пористая, и не будет оказывать существенного диффузионного сопротивления.
При величине КП-Б > 1 возможно образование плотной оболочки, когда КП-Б >> 1, возможно отслаивание твердой оболочки, в этом случае последняя не будет препятствовать диффузии.
Уравнение потока при наличии внутридиффузионных торможений имеет вид:  .
.
Признаки протекания процесса во внутренней диффузионной области:
1) Скорость процесса прямо пропорциональна концентрации реагента.
2) Скорость выщелачивания снижается по мере увеличения продолжительности процесса. Этот признак является определяющим.
3) Скорость сравнительно мало зависит от температуры. Значение энергии активации лежит в пределах 10 – 20 кДж/моль.
4) Скорость мало зависит от интенсивности перемешивания.
Закономерности и признаки кинетического режима выщелачивания. Химическая реакция - процесс, протекающий через ряд последовательных стадий. Скорость химической реакции подчиняется закону действия масс. При взаимодействии nA молекул вещества A c nB молекул вещества B скорость гомогенной реакции получения продукта D описывается уравнением:  , где G D - масса продукта; К – константа скорости реакции; СА и СВ – концентрации в растворе веществ А и В. А скорость гетерогенной:
, где G D - масса продукта; К – константа скорости реакции; СА и СВ – концентрации в растворе веществ А и В. А скорость гетерогенной:  .
.
Часто гетерогенные реакции протекают с участием реагентов, находящихся в адсорбированном состоянии. В этом случае реакции, приводящей к образованию конечного продукта, предшествует адсорбция растворенного реагента. В случае химической адсорбции адсорбированные молекулы связаны с поверхностью силами того же типа, что и силы, осуществляющие валентную связь. Химическая адсорбция требует энергии активации ("активированная" адсорбция). На скорость реакции оказывает влияние теплота адсорбции. Если молекулы в адсорбированном состоянии имеют меньшую энергию, чем в свободном, то избыток энергии активированного комплекса по отношению к исходному состоянию равен разности энергии активации и теплоты сорбции. В результате химическая адсорбция может оказаться более медленной стадией, чем последующая реакция, и тогда кинетическое уравнение гетерогенной реакции должно совпадать с кинетическим уравнением адсорбции.
Признаки протекания процесса выщелачивания в кинетическом режиме:
1) Уравнение потока выщелачивания в кинетической области имеет вид: 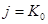 , из чего следует, что скорость находится в линейной зависимости от концентрации реагента.
, из чего следует, что скорость находится в линейной зависимости от концентрации реагента.
2) Скорость выщелачивания не зависит от интенсивности перемешивания.
3) Скорость процесса не зависит от его продолжительности при постоянных поверхности и концентрациях реагентов и продуктов реакции.
4) Скорость реакции, а значит и скорость выщелачивания, сильно зависят от температуры, с повышением температуры скорость химического взаимодействия резко возрастает. Энергия активации намного больше, чем для диффузионных режимов, и составляет 50–300 кДж/моль.
Методы активации процессов выщелачивания. Для процессов, протекающих в кинетическом режиме, основным фактором, влияющим на скорость процесса, является температура. Для интенсификации процессов выщелачивания используют некоторые специфические методы.
Механическая активация. Процесс связан с тонким измельчением исходной твердой фазы. При этом, происходит деформация и частичное разрушение кристаллической структуры твердой фазы, что приводит к уменьшению прочности этой фазы и, в конечном счете, к увеличению скорости ее взаимодействия с растворителем.
Возможны два варианта проведения процессов с механическим активированием:
1) Твердое тело сначала активируется в результате тонкого измельчения, затем производится выщелачивание растворителем;
2) Механическое воздействие (размол, истирание) совмещается с химическим взаимодействием с растворителем – механохимические процессы.
При механохимическом выщелачивании могут протекать реакции, которые в обычных условиях термодинамически маловероятны. Например, из термодинамических расчетов следует, что вода практически не реагирует с медью с выделением водорода. Однако при одновременном истирании в течение 5–6 дней около 2 % меди вступает в реакцию с водой с выделением водорода.
Термическая активация. Активирование твердых тел при термических воздействиях основано на уменьшении прочности связей в химических соединениях, нарушении кристаллической структуры, возникновении термических напряжений вследствие перепадов температуры, образовании более простых соединений, активных по отношению к растворителю. Это способствует нестабильности кристаллической структуры твердой фазы и более быстрому ее растворению.
Распространенным в металлургии процессом является активирующее термическое разложение сульфидов. Высшие сульфиды (пирит, арсенопирит, халькопирит) весьма трудно подвергаются выщелачиванию в неокисляющих минеральных кислотах. После термического разложения образуются простые сульфидные формы, хорошо растворимые в кислотах.
Биологическая активация (бактериальное выщелачивание). Микроорганизмы либо непосредственно воздействуют на сульфиды, либо ускоряют окисление ферросульфата. Наиболее распространенным типом бактерий, используемых в гидрометаллургии, являются Th. ferrooxidans. Для развития Th. ferrooxidans необходима кислая среда. Оптимальная величина рН в присутствии железа составляет 1,7–2,5; при рН > 6 бактерии не активны.
Механизм действия бактерий сводится к тому, что в процессе жизнедеятельности бактерии выделяют ферменты, являющиеся катализаторами протекающих реакций. Каталитическое действие фермента обусловлено активным центром его молекулы. Ферменты, присоединяясь с помощью активного центра к молекулам среды, образуют промежуточный активный комплекс, последующий распад которого дает конечные продукты. Благодаря образованию активного комплекса, снижается энергия активации реакции, и процесс выщелачивания идет с более высокой скоростью. Участие ферментов приводит к ускорению некоторых реакций в 10 9 – 10 14 раз.
Th.ferrooxidans выделяют ферменты, повышающие скорость окисления сульфата двухвалентного железа до сульфата трехвалентного железа. Последний, являясь сильным окислителем, выщелачивает сульфиды цветных металлов, например, меди. Образующийся при этом FeSO4 снова окисляется бактериями до Fe2(SO4)3.
Ультразвуковое активирование. Процессы выщелачивания интенсифицируются под воздействием ультразвукового поля (колебания с частотами выше 20 кгц). При распространении звуковой волны в жидкости возникают области переменного давления. В местах неоднородностей (у поверхности взвешенных твердых частиц) жидкость разрывается с образованием кавитационных полостей (каверн), которые в следующем полупериоде (сжатии) резко захлопываются. Газ в такой микрополости сжимается до нескольких тысяч атмосфер, затем происходит стремительное расширение и возникает ударная волна, которая производит разрушающее воздействие на твердую фазу: происходит измельчение твердых частиц, разрушение кристаллов, снятие поверхностных пленок. Кроме того, возникают гидродинамические потоки вихревого характера, способствующие уменьшению внешнедиффузионных сопротивлений. Все это приводит к резкому увеличению скорости гетерогенного процесса выщелачивания.
 Механизм и кинетика цементации. Цементация – это электрохимический процесс, описываемый реакцией: Z2Me1Z1+ Z1Me20 = Z2 Me10 + Z1Me2Z2, где Z1 и Z2 – заряды ионов.
Механизм и кинетика цементации. Цементация – это электрохимический процесс, описываемый реакцией: Z2Me1Z1+ Z1Me20 = Z2 Me10 + Z1Me2Z2, где Z1 и Z2 – заряды ионов.
При погружении металла – цементатора (Ме2) в раствор, содержащий ионы металла (Me1Z+) начинается взаимодействие, в результате которого на поверхности Ме2 образуются катодные участки, покрытые вытесняемым металлом. Одновременно возникают анодные участки, где протекает процесс ионизации атомов вытесняющего металла (растворение Ме2) (рисунок 4).
Возникновение катодных и анодных участков на поверхности металла цементатора обусловлено различным уровнем энергии в различных точках поверхности. Катодные участки будут возникать в местах поверхности с более высоким электродным потенциалом. Поскольку катодные и анодные участки соединены, электроны перетекают от анодных участков к катодным, где происходит разряд ионов вытесняемого металла (внутренняя цепь). Внешней цепью такого короткозамкнутого элемента является электролит, омическое сопротивление которого зависит от концентрации ионов в растворе.
Таким образом, цементация состоит из 2-х последовательных стадий:
1) Диффузионные процессы доставки ионов к катодной поверхности и отвода ионов от анодной поверхности.
2) Электрохимические превращения, т.е. разряд ионов на катодных участках и ионизация металла на анодных участках.
Катодный процесс включает дегидратацию иона, сорбцию его на поверхности, разряд иона с образованием атома металла, внедрение его в кристаллическую решетку цементируемого металла. Анодный процесс: ионизация атома (образованием иона) и его десорбция. Скорость цементации определяется скоростью самой медленной (лимитирующей) стадии, которая зависит от величины и характера электродной поляризации. Поляризация - смещение потенциала электрода от равновесного значения, происходящее при прохождении тока через гальванический элемент. Явление поляризации объясняется тем, что отвод электронов от анода и приток их к катоду совершается со значительно большей скоростью, чем электродные реакции (разряд ионов на катоде или ионизация металла на аноде) и диффузионные процессы подвода и отвода ионов. При катодной поляризации подвод электронов к катодным участкам опережает скорость подвода и разряда катионов, накопление отрицательных зарядов смещает потенциал в отрицательную сторону. При анодной поляризации скорость образования и отвода ионов в раствор меньше скорости отвода электронов, что приводит к накоплению избыточных положительных зарядов и смещению потенциала в положительную сторону.
Различают концентрационную (диффузионную) поляризацию, вызванную малой скоростью доставки (отвода) ионов по сравнению со скоростью их разряда (образования), и химическую поляризацию, обусловленную меньшей скоростью разряда ионов (или ионизации атомов) по сравнению со скоростью доставки их к поверхности электрода (или отвода от нее).
В практике известно много случаев катодного и анодного ограничения процесса цементации. Так, установлено, что цементацию меди на никеле лимитирует ионизация никеля на аноде. При цементации же меди на цинке и кадмии процесс контролируется разрядом ионов меди на катодных участках.
Диффузионные ограничения катодного процесса обычно проявляются в конце процесса цементации при малой концентрации ионов в растворе.
Диффузионное торможение на анодных участках может возникнуть при образовании в процессе цементации относительно толстого слоя цементируемого металла, через поры которого должны диффундировать ионы в объем раствора, однако этот случай редко встречается в практической гидрометаллургии.
 В общем виде зависимость скорости цементации от времени приведена на рисунке 5. В начальный период скорость возрастает со временем, что соответствует формированию и развитию катодных участков. Далее скорость цементации постепенно снижается по мере уменьшения концентрации цементируемого металла и сокращения анодной поверхности.
В общем виде зависимость скорости цементации от времени приведена на рисунке 5. В начальный период скорость возрастает со временем, что соответствует формированию и развитию катодных участков. Далее скорость цементации постепенно снижается по мере уменьшения концентрации цементируемого металла и сокращения анодной поверхности.
Повышение температуры не всегда увеличивает скорость цементации. Так, увеличение температуры ускоряет цементацию меди на цинке, но ухудшает осаждение кадмия на цинке.
При повышенных температурах происходит обратное химическое растворение кадмия в слабокислых растворах.
Скорость цементации зависит от величины удельной поверхности и активности цементирующего металла. Например, цинковая пыль, полученная конденсацией паров цинка, активней пыли, полученной распылением жидкого цинка струей сжатого воздуха.
Поиск по сайту: